Hi, I am trying to run lammps on windows in parallel, and I followed instructions provided by https://docs.lammps.org/Run_windows.html
I succeeded in running sample from lammps examples (in.melt.lammps), but when I tried to run my own input files, it doesnt work and error below shown:
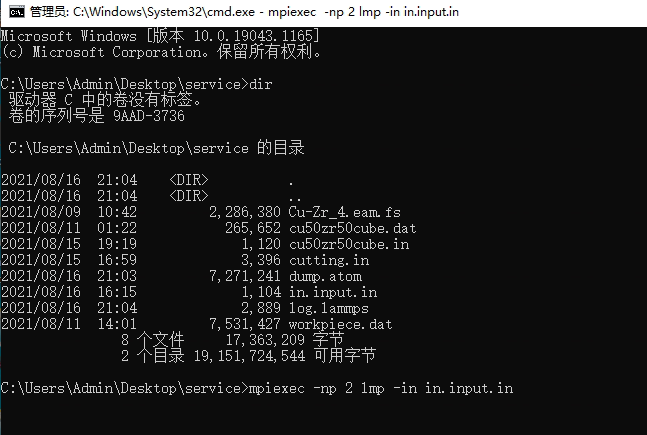
ERROR on proc 0: Cannot open input script in.file: No such file or directory (src/lammps.cpp:508)
What I entered: mpiexec -np 2 lmp -in in.file
It would be great if anyone could provide some guidance. Thank you in advance.
Please make certain that you really have a file called in.file in the folder where you are running LAMMPS, because LAMMPS cannot obviously find it.
Best to check from the command line with dir. The file names in the file browser may be misleading since by default windows will remove known file extensions that are associated with programs (like .txt).
You probably didn’t do all the required setup steps. You may also need to modify your firewall settings.
Also consider using localonly instead of np. There is a summary of how to setup and use MPICH on windows on the LAMMPS/windows website, but for details you need to consult the documentation for the specific MPICH version. Some steps are different from the usual Linux behavior.
@akohlmey I tested each method u mentioned. Firewall and localonly werent the problem, so I guess I can only search for more info online. Anyways, thank you very much.
I know why: output buffering. Output will only appear when there is more than 4k characters to show and then it will output those 4k and stall until the next 4k have accumulated or the program has ended.
This is a “feature” of the OS and the MPI library and there is nothing that can be done about it from the LAMMPS side.