Dear developers and user,
I’m running the simulation using an unmodified LAMMPS (2 Aug 2023) version. I want to use multiple GPUs and CUDA API to accelerate the PPPM alogrithm. However, I can’t access more than one GPU when running program. I have tried numerous ways to solve this problem, but I have not succeeded yet.
Hardware and Software Platform
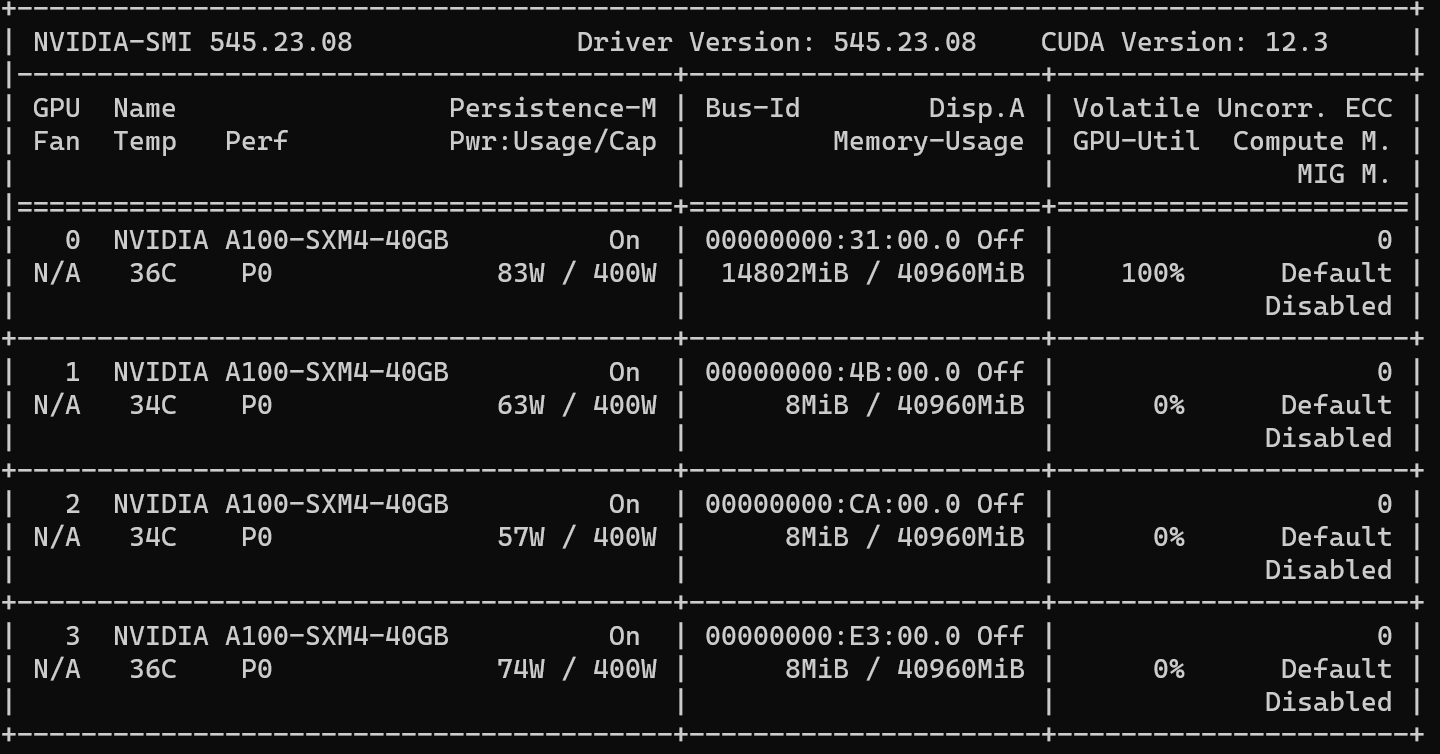
My sever has 64 CPUs and 4 Nvidia A100 GPUs whose hardware configuration is below:
+---------------------------------------------------------------------------------------+
| NVIDIA-SMI 545.23.08 Driver Version: 545.23.08 CUDA Version: 12.3 |
|-----------------------------------------+----------------------+----------------------+
| GPU Name Persistence-M | Bus-Id Disp.A | Volatile Uncorr. ECC |
| Fan Temp Perf Pwr:Usage/Cap | Memory-Usage | GPU-Util Compute M. |
| | | MIG M. |
|=========================================+======================+======================|
| 0 NVIDIA A100-SXM4-40GB On | 00000000:31:00.0 Off | 0 |
| N/A 45C P0 90W / 400W | 7405MiB / 40960MiB | 100% Default |
| | | Disabled |
+-----------------------------------------+----------------------+----------------------+
| 1 NVIDIA A100-SXM4-40GB On | 00000000:4B:00.0 Off | 0 |
| N/A 43C P0 54W / 400W | 8MiB / 40960MiB | 0% Default |
| | | Disabled |
+-----------------------------------------+----------------------+----------------------+
| 2 NVIDIA A100-SXM4-40GB On | 00000000:CA:00.0 Off | 0 |
| N/A 42C P0 55W / 400W | 8MiB / 40960MiB | 0% Default |
| | | Disabled |
+-----------------------------------------+----------------------+----------------------+
| 3 NVIDIA A100-SXM4-40GB On | 00000000:E3:00.0 Off | 0 |
| N/A 44C P0 63W / 400W | 8MiB / 40960MiB | 0% Default |
| | | Disabled |
+-----------------------------------------+----------------------+----------------------+
And here are libraries I use:
- cmake/3.26.3-intel-2021.4.0
- cuda/12.1.1
- oneapi/2021.4.0
- intel-oneapi-mpi/2021.4.0
- intel-oneapi-mkl/2021.4.0
- intel-oneapi-tbb/2021.4.0
- intel-oneapi-compilers/2021.4.0
Compile, Launch and Output
Here are my compiling commands:
$ cmake -C ../cmake/presets/most.cmake -C ../cmake/presets/nolib.cmake -D PKG_GPU=on -D GPU_API=cuda -D LAMMPS_MACHINE=pppm_gpu ../cmake
$ cmake --build ..
And here is my launch command:
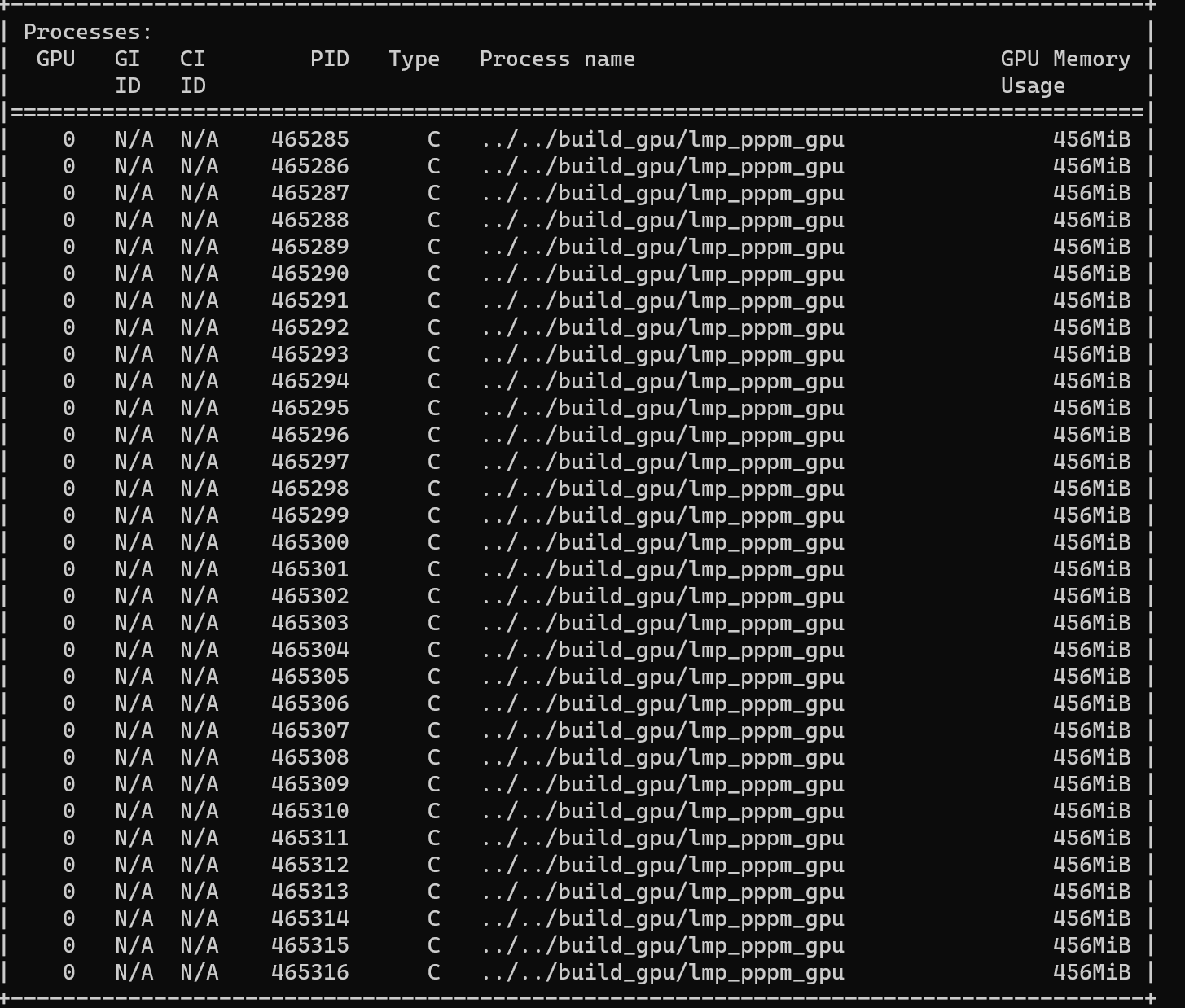
mpirun -n 2 ../../build_gpu/lmp_pppm_gpu -sf gpu -in in.spce-bulk-nvt
And this is my input file in.spce-bulk-nvt:
# SPC/E water box bulk
log PPPM500GPU2-2.out
package gpu 2 device_type nvidiagpu omp 2
units real
atom_style full
read_data equi_bulk.4000000.data
group O type 1
group H type 2
group water type 1:2:1
replicate 1 1 1
pair_style lj/cut/coul/long 10.0 10.0
kspace_style pppm/gpu 0.071
#kspace_style pppm 0.071
pair_coeff 1 1 0.1556 3.166
pair_coeff * 2 0.0000 0.0000
bond_style harmonic
angle_style harmonic
bond_coeff 1 1000.00 1.000
angle_coeff 1 100.0 109.47
special_bonds lj/coul 0 0 0.5
neighbor 2.0 bin
neigh_modify every 10 delay 10 check yes one 5000
thermo_style custom step etotal temp
thermo_modify line one
thermo 100
#===================================================
fix 1 water shake 0.0001 5000 0 b 1 a 1
fix 2 water nvt temp 298 298 5
timestep 1
run 100000
write_data equi_bulk.*.data nocoeff
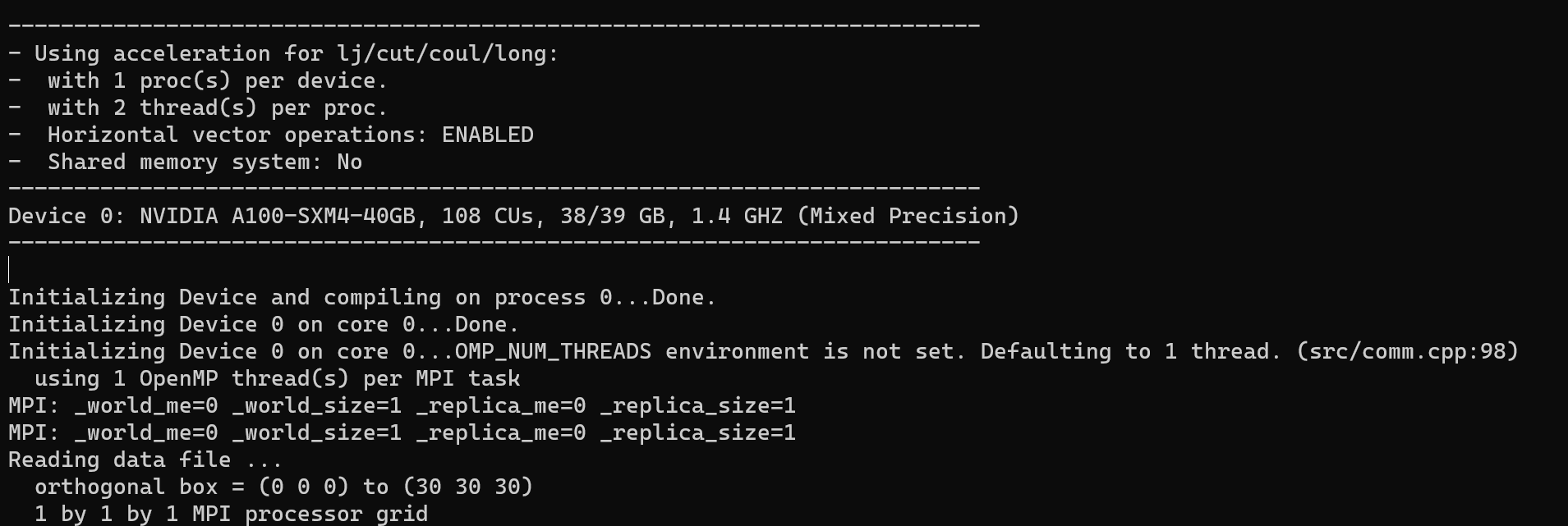
And here is the output initialization information( I printed additional variables, including NodeRank , Proc(s) per node , FirstGPU , LastGPU , and Devices Number .):
PPPM initialization ...
using 12-bit tables for long-range coulomb (src/kspace.cpp:342)
G vector (1/distance) = 0.04463336
grid = 1 1 1
stencil order = 5
estimated absolute RMS force accuracy = 27.659355
estimated relative force accuracy = 0.083295326
using double precision KISS FFT
3d grid and FFT values/proc = 216 1
--------------------------------------------------------------------------
- NodeRank: 0
- Proc(s) per node = 1
- FirstGPU: 0
- LastGPU: 0
- Devices Number: 1
- Using acceleration for pppm:
- with 1 proc(s) per device.
- with 2 thread(s) per proc.
- Horizontal vector operations: ENABLED
- Shared memory system: No
--------------------------------------------------------------------------
Device 0: NVIDIA A100-SXM4-40GB, 108 CUs, 39/39 GB, 1.4 GHZ (Mixed Precision)
--------------------------------------------------------------------------
My Analysis
I tried to analyze this problem in three aspects:
- Hardware Platform
- Software Dependencies
- Launch Commands
And I explain my analysis from above three aspects.
Hardware
I print the GPUs’ device information by a simple CUDA program. And the 4 GPUs can be detected by program correctly.
What’s more, I can also change the device ID by using the command:
package gpu 2 gpuID 1
In this case, the program uses device 1 to run the kernel function.
Software Dependencies
I haven’t a good idea to verify whether the software dependencies are correct. However, I remember that about a month ago, when I last used those dependencies, the PPPM’s GPU version could correctly utilize multiple GPUs.
Launch Commands
After all the above efforts, I believe I need to examine the device codes to understand how the GPU devices are allocated. The code file is in .\lib\gpu\lal_device.cpp.
/****Codes in `.\lib\gpu\lal_device.cpp`****/
// Get the rank/size within the world
MPI_Comm_rank(_comm_world,&_world_me);
MPI_Comm_size(_comm_world,&_world_size);
// Get the rank/size within the replica
MPI_Comm_rank(_comm_replica,&_replica_me);
MPI_Comm_size(_comm_replica,&_replica_size);
// Get the names of all nodes
int name_length;
char node_name[MPI_MAX_PROCESSOR_NAME];
auto node_names = new char[MPI_MAX_PROCESSOR_NAME*_world_size];
MPI_Get_processor_name(node_name,&name_length);
MPI_Allgather(&node_name,MPI_MAX_PROCESSOR_NAME,MPI_CHAR,&node_names[0],
MPI_MAX_PROCESSOR_NAME,MPI_CHAR,_comm_world);
std::string node_string=std::string(node_name);
// Get the number of procs per node
std::map<std::string,int> name_map;
std::map<std::string,int>::iterator np;
for (int i=0; i<_world_size; i++) {
std::string i_string=std::string(&node_names[i*MPI_MAX_PROCESSOR_NAME]);
np=name_map.find(i_string);
if (np==name_map.end())
name_map[i_string]=1;
else
np->second++;
}
int procs_per_node=name_map.begin()->second;
_procs_per_node = procs_per_node;
// Assign a unique id to each node
int split_num=0, split_id=0;
for (np=name_map.begin(); np!=name_map.end(); ++np) {
if (np->first==node_string)
split_id=split_num;
split_num++;
}
delete[] node_names;
// Set up a per node communicator and find rank within
MPI_Comm node_comm;
MPI_Comm_split(_comm_world, split_id, 0, &node_comm);
int node_rank;
MPI_Comm_rank(node_comm,&node_rank);
// ------------------- Device selection parameters----------------------
if (ndevices > procs_per_node)
ndevices = procs_per_node;
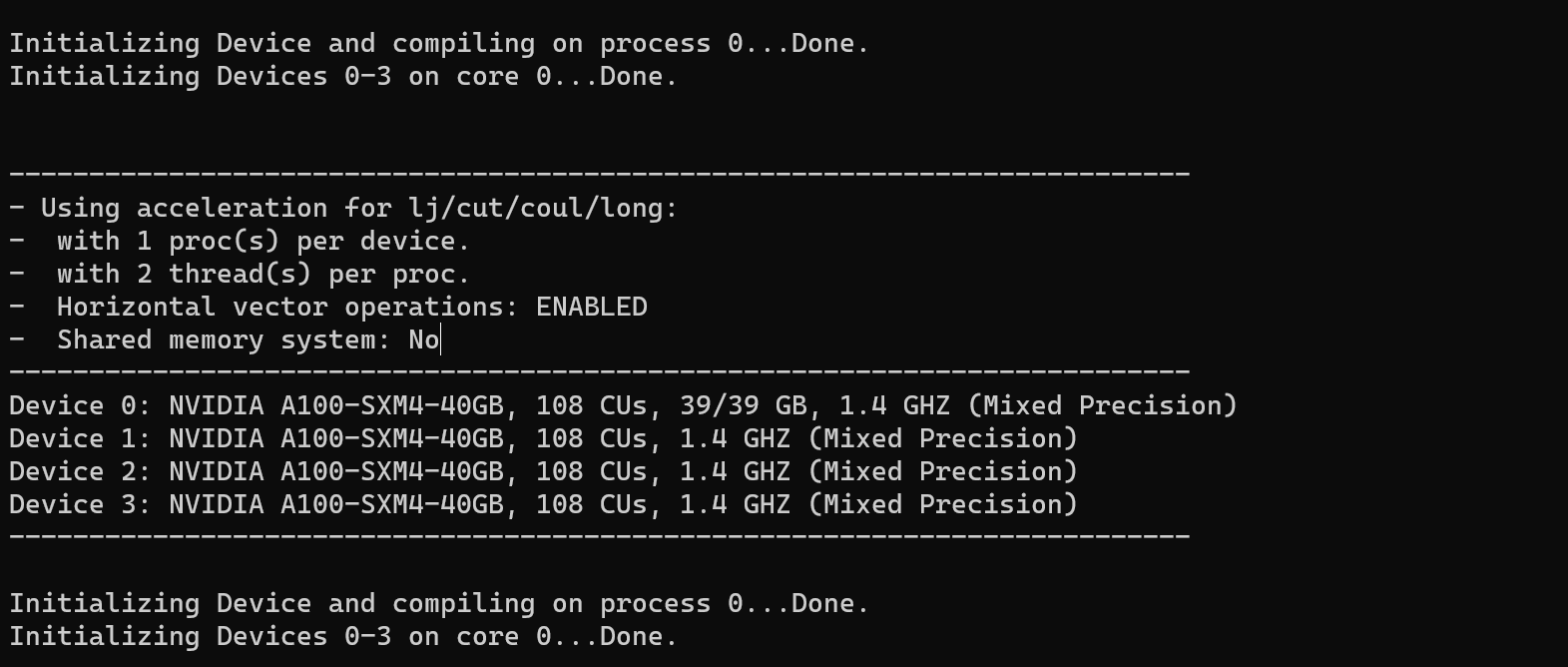
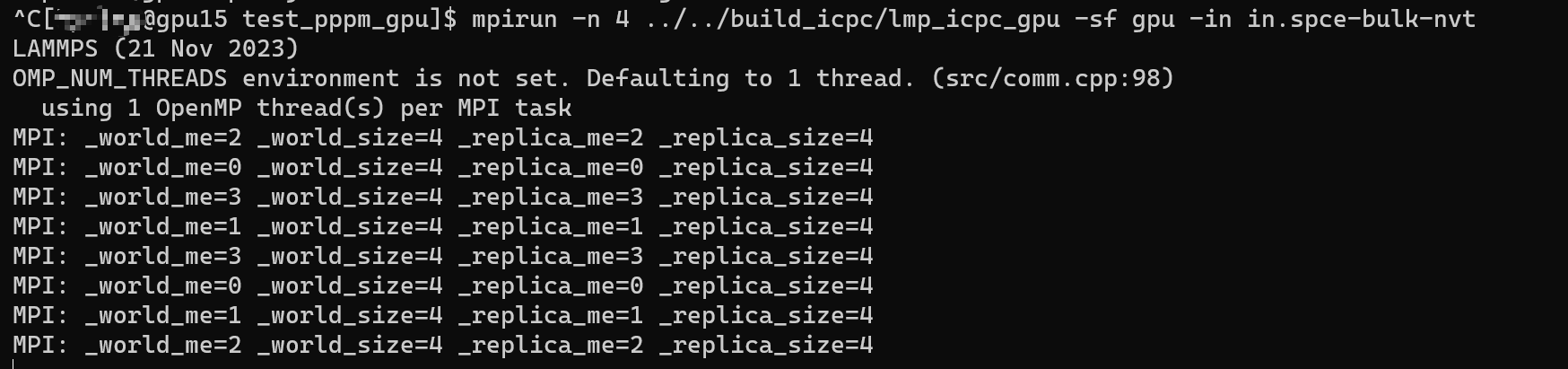
After reading through the codes and printing key variables, I’ve identified two problems that I believe may be crucial to solving the entire issue.
- The MPI’s world rank,
_world_me, is always 0, which leads that all the MPI processes use the same GPU device. - The variable
procs_per_node, which represents the number of procs per node, is always 1, resulting in the program using only one GPU regardless of the number specified in the input command.
I have no idea how to solve those problems. I’ve tried many different mpirun -n and -ppn combinations, but I’m still facing challenges. Could someone please offer guidance on my issue? I’m particularly struggling. Any suggestions, explanations, or examples would be greatly appreciated. Thank you in advance for your help!