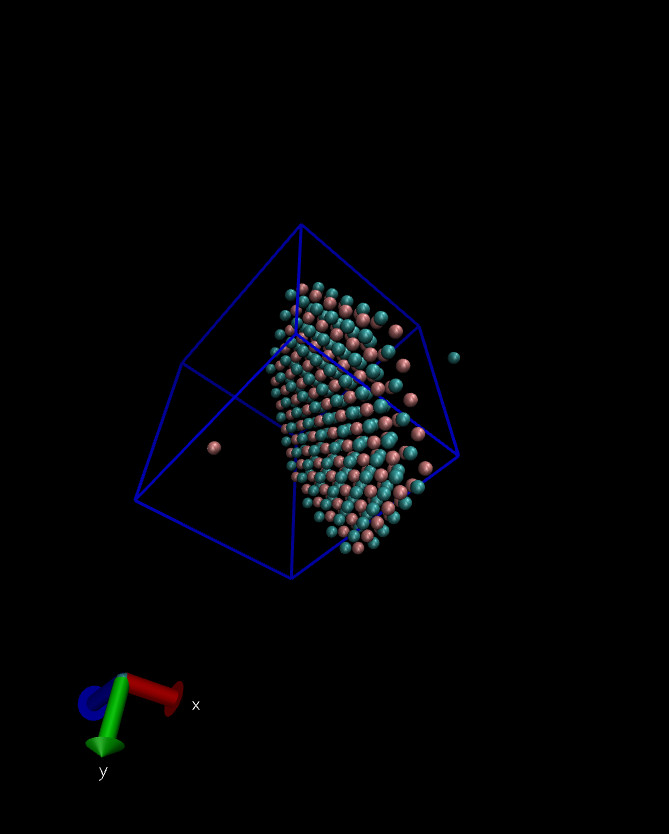
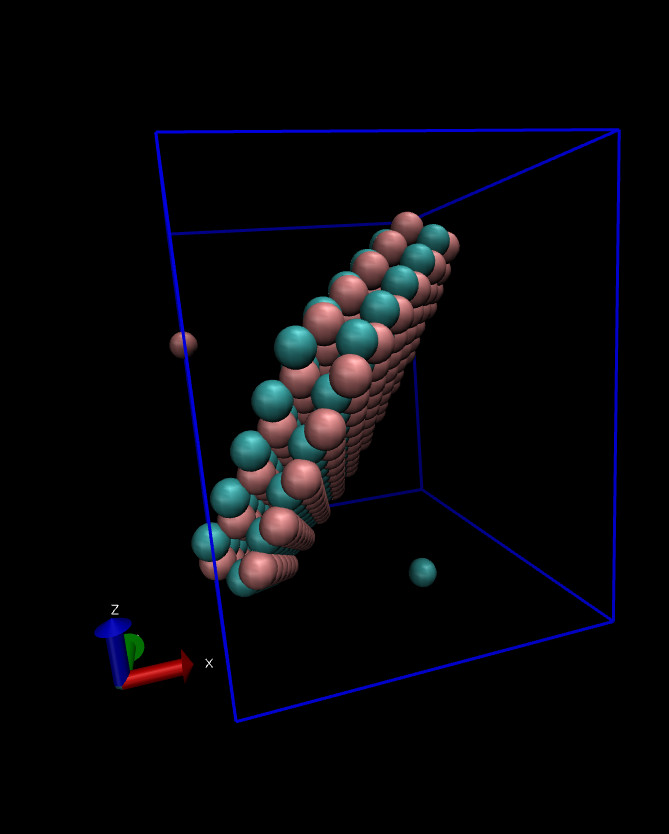
Hi,
I have a PDB file that I want to convert to a LAMMPS data file format. I tried saving the coordinates using VMD and exporting them as a lammpstrj file, but the resulting axis lo and hi values are incorrect (from -0.5 to +0.5). When I use the command topo writelammpsdata lmp.data full, the box dimensions are set to:
X-axis: 2.639 to 30.334
Y-axis: 7.679 to 42.551
Z-axis: -5.921 to 28.597
I would like to center this slab within the box so that there are no negative axis values. The issue arises during the simulation when two particles move outside the box. How can I adjust the coordinates to center the slab and avoid negative axis values?
I am attaching the snapshot of the pdb and after simulation
The PDB file does not have box information, so you would have to set the box dimensions from the VMD command prompt manually.
If there is no box given, TopoTools will use the min/max of the positions to determine the box.
If you set the box dimensions manually, TopoTools will use that information. But since VMD doesn’t store the box origin (only the extent) the box will be centered around the positions.
You have multiple options:
you can manually edit the box dimension in the files (this will not translate the positions) and then use the displace_atoms command in LAMMPS (this will only work for periodic boundaries)
you can set the box dimensions in VMD before saving
you can move the atoms to the center in VMD first and then set the box dimensions
Hi,
I displaced the initial structure using the displace_atoms command with the following dimensions:
0 27.695 xlo xhi
0 34.872 ylo yhi
0 34.518 zlo zhi
I saved the results in final_topo.pdb and final_topo.data files. When visualizing, final_topo.pdb displayed the correct structure. However, after running the simulation, the output dump trajectory shows two particles outside the slab (see figure below)
and with shifted coordinates. There should not be any shift of coordinates, that must remain same as the final_topo.data as the coordinates are frozen. I’m unsure why this discrepancy is occurring.
My Lammps script:
units real
dimension 3
boundary p p p
atom_style full
Please have a look at the forum guidelines for how to correctly quote text file. Can you read everything correctly at the moment?
It is difficult to tell without the actual files, but if the atoms are at the corner of the box, just the slightest numerical noise can move those atoms to the next periodic box. That is not an error. If you want those atoms to “stick together” just output unwrapped coordinates.
The best way to keep atoms immobile is to not include them in the time integration. That also avoids incorrect thermostatting.
There don’t seem to be any mobile atoms in your system, so why running an MD, why using a thermostat?
many thanks for the suggestions. Actually, I’m supposed to incorporate free random ions in the box. Anyway, what you say is right, bcz for initial check it is not required. displace_atoms helped.