Hello,
I am calculating the twinning fault energy of a number of EAM potentials for FCC metals (from this site https://sites.google.com/site/eampotentials/).
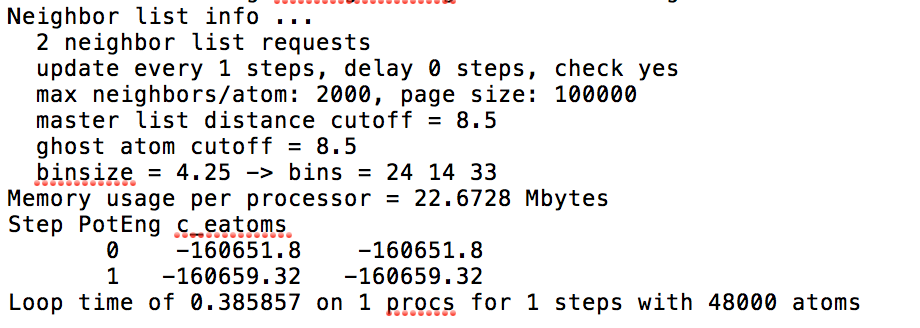
I have a simple script that automatically generates perfect, oriented FCC crystals by stacking up two half crystals (defined by regions), and minimizes the energy of the structure via cg. Dump files before minimization show that a perfect crystal is generated in each case (boundary conditions are p p f). However, during minimization, nan values are returned for certain potentials. I attach a “good” and “bad” case below. Note that the potential calls are correct and used in other scripts. My question is, what could be causing the nan values during minimization?
I have read that “bad” structures lead to NaN values, but dumping them beforehand shows that the structures are as intended. Could it be numerical instability of the dimensions input to the region command that are causing these nan values? I am using variables as the box dimensions input to the region command. I have found in other scripts for the potentials that are giving NaN below that explicitly entering numbers into the region command rather than variables defined as those numbers does not produce this issue. Any insight into why nan values are being produced is welcome. See log file attached as well.
Thank you for the help,
log_success_GTFE.txt (111 KB)
log_failure_GTFE.txt (7.02 KB)
Hello,
I am calculating the twinning fault energy of a number of EAM potentials
for FCC metals (from this site https://sites.google.com/
site/eampotentials/).
I have a simple script that automatically generates perfect, oriented FCC
crystals by stacking up two half crystals (defined by regions), and
minimizes the energy of the structure via cg. Dump files before
minimization show that a perfect crystal is generated in each case
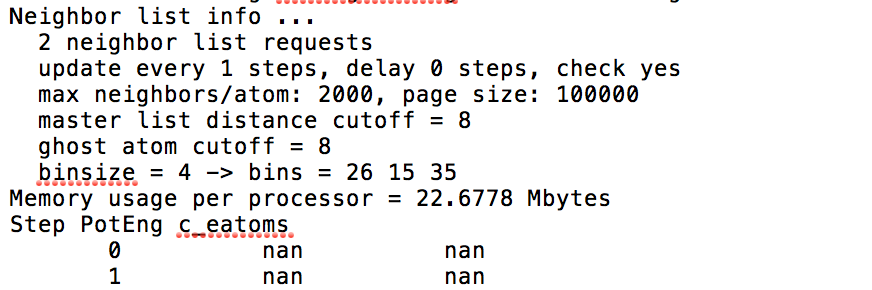
(boundary conditions are p p f). However, during minimization, nan values
are returned for certain potentials. I attach a "good" and "bad" case
below. Note that the potential calls are correct and used in other scripts.
My question is, what could be causing the nan values during minimization?
I have read that "bad" structures lead to NaN values, but dumping them
beforehand shows that the structures are as intended. Could it be numerical
instability of the dimensions input to the region command that are causing
these nan values? I am using variables as the box dimensions input to the
region command. I have found in other scripts for the potentials that are
giving NaN below that explicitly entering numbers into the region command
rather than variables defined as those numbers does not produce this issue.
Any insight into why nan values are being produced is welcome. See log file
attached as well.
rounding can be the origin for your issues. visual inspection is not
always sufficient to tell if you have close contacts.
a simple test would be to use the delete_atoms with the overlap option
and a suitable cutoff, to identify overlapping atoms.
axel.