Dear All,
I did two consecutive simulations: equilibration followed by shock propagation.
(1) in equilibration, I used periodic conditions p p p, and pair_style lj/charmm/coul/long and kspace_style pppm.
at the end of equilibration, some atoms are outside of the local cell.
(2) for the shock simulation (using “read_restart” the restart file saved in equilibration), I have to change the periodic boundary conditions to non-periodic : s p p by using “change_box.”
But I got a warning and an error: WARNING: Bond/angle/dihedral extent > half of periodic box length (…/domain.cpp:680)
ERROR on proc 2: Bond atoms 15495 15497 missing on proc 2 at step 94 (…/neigh_bond.cpp:55)
My question is: Does Lammps still take account the atoms that are outside of the local cell when I change the boundary from “p” to “s”?
I observed the dimensions of the local cell are the same between the last step of equilibration (p p p) and the first step of the shock simulation (s p p).
Thank you very much for your help,
Lili
Dear All,
I did two consecutive simulations: equilibration followed by shock
propagation.
(1) in equilibration, I used periodic conditions *p p p*, and *
pair_style lj/charmm/coul/long *and* kspace_style pppm*.
at the end of equilibration, some atoms are outside of the local cell.
This equilibration is not complete. If you change the boundary to s p p
you are creating two free surfaces that should be further equlibrated
before moving on to shock simulatons - so you are missing a step here.
To prevent atoms passing the boundary during equilibrations, from the start
of the equilibration you can change the boundary to s p p and add a
wall/reflect on xlo and xhi edges. This way you equilibrate the surfaces
at the same time and it will prevent the errors you saw from happening.
Ray
Dear All,
I did two consecutive simulations: equilibration followed by shock propagation.
(1) in equilibration, I used periodic conditions p p p, and pair_style lj/charmm/coul/long and kspace_style pppm.
at the end of equilibration, some atoms are outside of the local cell.
This equilibration is not complete. If you change the boundary to s p p you are creating two free surfaces that should be further equlibrated before moving on to shock simulatons - so you are missing a step here.
To prevent atoms passing the boundary during equilibrations, from the start of the equilibration you can change the boundary to s p p and add a wall/reflect on xlo and xhi edges. This way you equilibrate the surfaces at the same time and it will prevent the errors you saw from happening.
Or you can use two instances of fix oneway with opposite sign to separate the bulk system before changing the boundary periodicity. Fix oneway is similar to wall/reflect only that it is compatible with periodic boundaries.
Axel
Dear Axel and Ray,
Thanks for your guidance as always. Yes, I missed a further equilibration after changing boundaries from “p p p” to “s p p”.
But I am having a conceptual confusion on the new surfaces I created, please see the figure attached (I just draw x dimension for simplicity):
(0) I started with a bio-molecules system under periodic conditions p p p.
(1) I equilibrated the system under p p p, and at the end the equilibration some atoms move outside of the local cell.
(2) I used “change_box” to change boundary from “p p p” to “s p p,” and I am not very sure which system I will get: Fig A or FigB ??
If it is like Fig A, then the surface I created passing through the molecules.
If it is like Fig B, then I don’t get correct bonding connectivity, for example atom2 and atom3.
Am I misunderstanding something on the doc page?
Thank you very much,
Lili
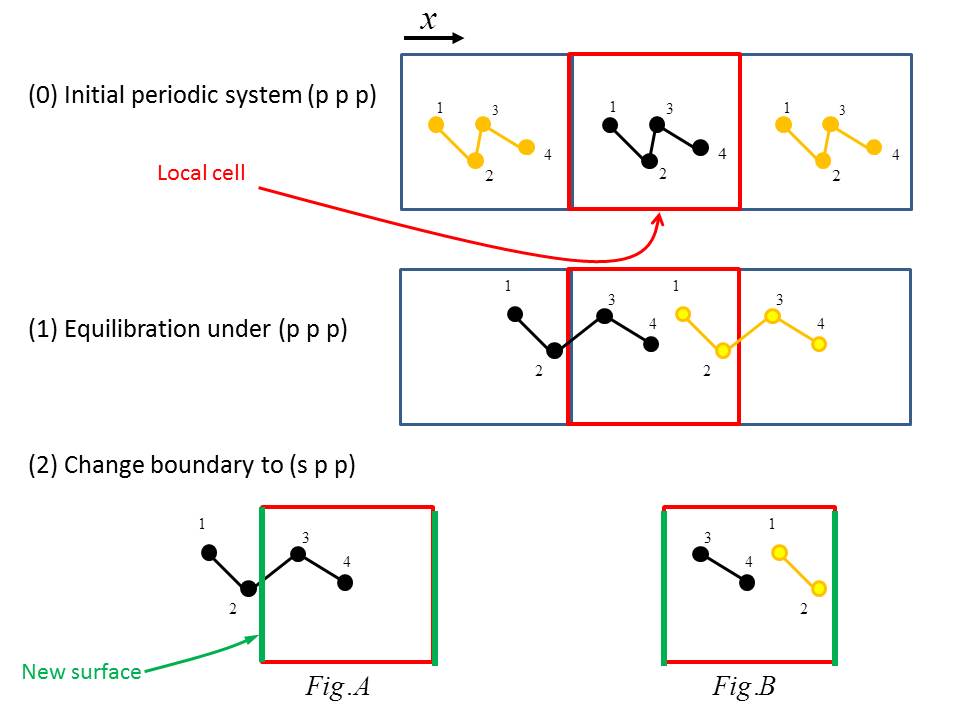
Dear Axel and Ray,
Thanks for your guidance as always. Yes, I missed a further equilibration
after changing boundaries from "p p p" to "s p p".
But I am having a conceptual confusion on the new surfaces I created, please
see the figure attached (I just draw x dimension for simplicity):
(0) I started with a bio-molecules system under periodic conditions p p p.
(1) I equilibrated the system under p p p, and at the end the equilibration
some atoms move outside of the local cell.
(2) I used "change_box" to change boundary from "p p p" to "s p p," and I am
not very sure which system I will get: Fig A or FigB ??
neither would be desirable; hence the suggestions to either start with
reflecting walls and s p p from the beginning or use fix oneway to
create a little void around the plane, where you want to cut the
periodicity, so that you don't have to worry about bonds crossing the
periodic boundary in the first place.
axel.