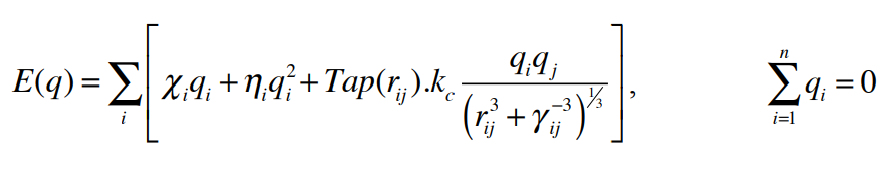Hello,
I have a question and I would be thankful if you could help me.
I am trying to change a small part of the Lammps source code to add an Electric Field parameters (ex, ey, ez) to the reaxff_nonbonded.cpp and fix_qeq_reaxff files. These changes solve the problem of unphysical long-range charge transfer which have seen in qeq method. I could add efield to the fix_qeq_reaxff file successfully, but I got some errors for the reaxff_nonbonded.cpp file. What I did was add these command lines to the reaxff_nonbonded.cpp file:
double x, y , z;
double ex_mtn = system->efield->ex;
double ey_mtn = system->efield->ey;
double ez_mtn = system->efield->ez;
x = system->my_atoms[i].x[0];
y = system->my_atoms[i].x[1];
z = system->my_atoms[i].x[2];
en_tmp = KCALpMOL_to_EV * ((system->reax_param.sbp[type_i].chi * q +(system->reax_param.sbp[type_i].eta / 2.) * SQR(q))-q*(xex_mtn+yey_mtn+z*ez_mtn));
In addition, I added " class FixEfield *efield; " in “structure reax_system {}” section of the “reaxff_type.h” file.
I attached the changed files. In these files, I have shown added lines by "//…nina " mark.
I got the error: invalid use of incomplete type ‘class ReaxFF::FixEfield’.
I tried different things but none of them worked. I will be thankful if anyone can help me solve this error.
reaxff_types.h (8.2 KB)
reaxff_nonbonded.cpp (20.5 KB)
fix_qeq_reaxff.cpp (30.3 KB)