Hello
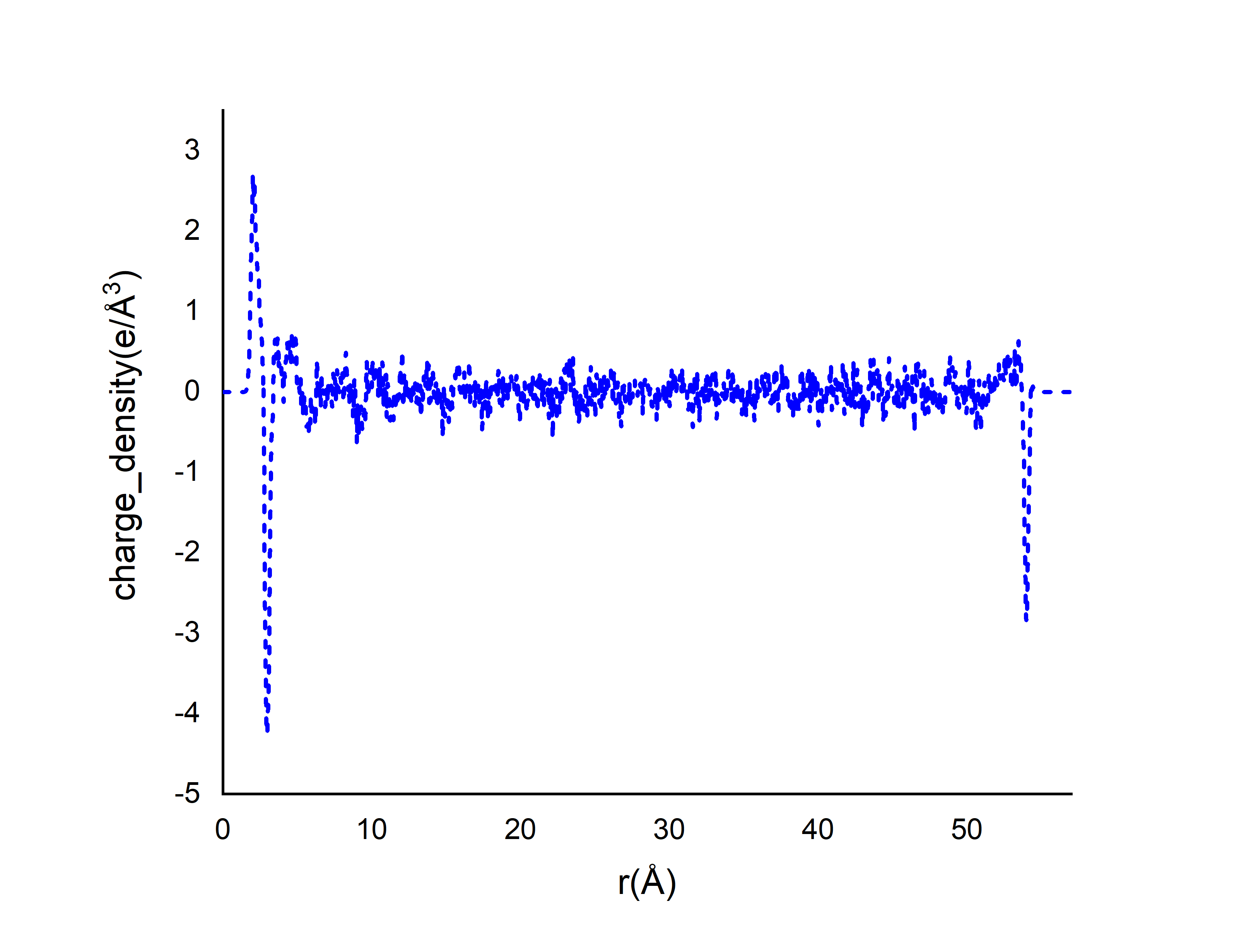
I computed the charge density profile along z using some commands in lammps. and I want to use these values to calculate electric potential profile. My simulation box contains graphene electrodes and NaCl(2M) electrolyte.
In some papers the range of charge density for (water+NaCl electrolyte system) is from -0.1 to +0.1 e/Å3) but my charge density value is very big.
I used this commands:
compute charge1 all property/atom q
compute C all chunk/atom bin/1d z lower 0.05
fix 81f all ave/chunk 100 100 10000 C c_charge1 norm none file charge.profile
and I plotted it:
This is not really a LAMMPS issue but a question of the science that you are trying to model. The profile looks like that of a system where atoms/molecules are forming layers at surfaces.
You should discuss this with your adviser/supervisor/tutor.
Do you want 0.05-angstrom bins (which is, I think, what the default will end up being, since the default for chunk/atom is units lattice and the default for lattice is 1)?
If yes, then this is expectable. Your big peaks look like the graphene sheets (where, since all the charges are in the same z layer, the charge density will depend on bin size) and you have enough noise in between to look like 0.05 angstrom bins.
If no, and what you really wanted was 20 bins throughout the box, then you need units reduced for your chunk/atom command.