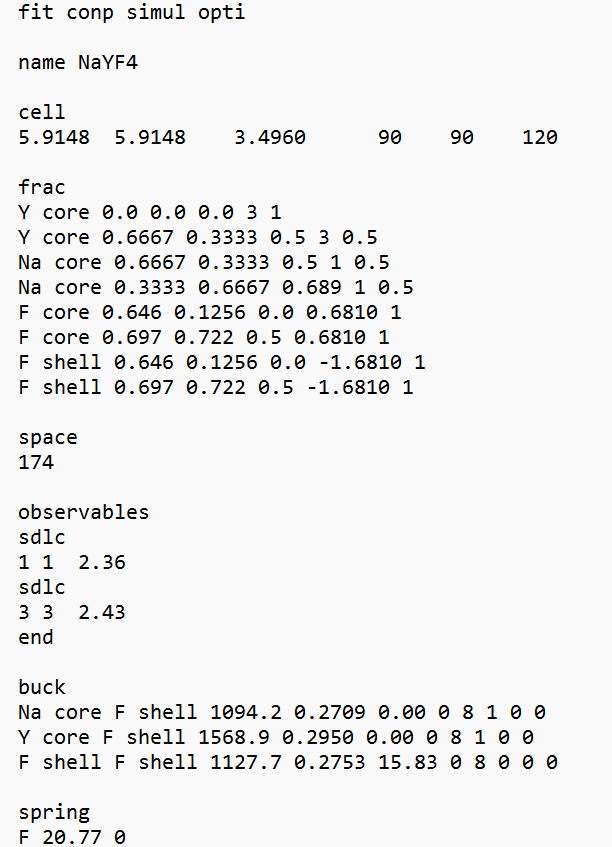
I am trying to find Interatomic potential (Buckingham potential) for hexagonal phase of NaYF₄ in GULP 6.2 (running via Cygwin on Windows 11).
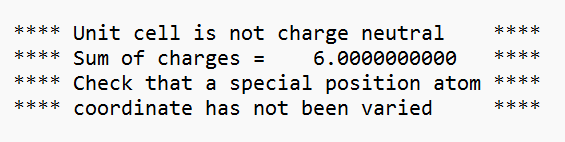
I am encountering an error stating that the unit cell is not charge neutral.
What could be causing this discrepancy? Are there specific aspects of charge assignment, potential parameters, or computational settings that might lead to this issue in GULP?
Would using the qok keyword be appropriate in this case?
I have attached snippet of input file.
I have attached snippet of the error message.
Any guidance would be greatly appreciated.
Best regards,
Aravind