Hi,
I am trying to deposit TiO2 on Si substrate. ( simulate a magnetron sputtering on Si substrate)
First, I have first formed Si substrate found the equib lattice via NPT runs, removed half plane in the top surface and added 10A buffer zones to the data file in both z directions, removed the periodicity in z in the input script and ran it for NVT. Now I have the Si surface, but I used atomic style for generation of the surface so my current data file is in atomic format. Now in magnetron sputtering particles are charged so I have to include that also.
Data File
LAMMPS data file via write_data, version 29 Sep 2021, timestep = 3100000
7303 atoms
3 atom types
0.01887559500250191 54.25337321499684 xlo xhi
0.01887559500250191 54.25337321499684 ylo yhi
-10.00000 65.00000 zlo zhi
Masses
1 28.0855
2 47.867
3 16.000
Atoms # atomic
1 1 1.3407728263786889 1.3583167141708739 1.3925263426717569 0 0 0
2 1 6.7274841734057009 1.3185113025147779 1.3931498266753020 0 0 0
3 1 8.1276281904510981 0.1533973093788366 2.7763599479603318 0 0 0
4 1 10.7926763723013419 2.7796469604562137 2.7725943886935775 0 0 0
----------
I have two questions:
1) My data file initially had 1 atom type so I added 2 and 3 for Ti and O later before I start the deposition. How can I start a charged particle deposition on this existing data file? what is the input stricpt modification to address this change? Otherwise deposition of Ti and O directly will be just neutral particles falling down right? I think I also have to define atomic charges for Ti and O explicitly.
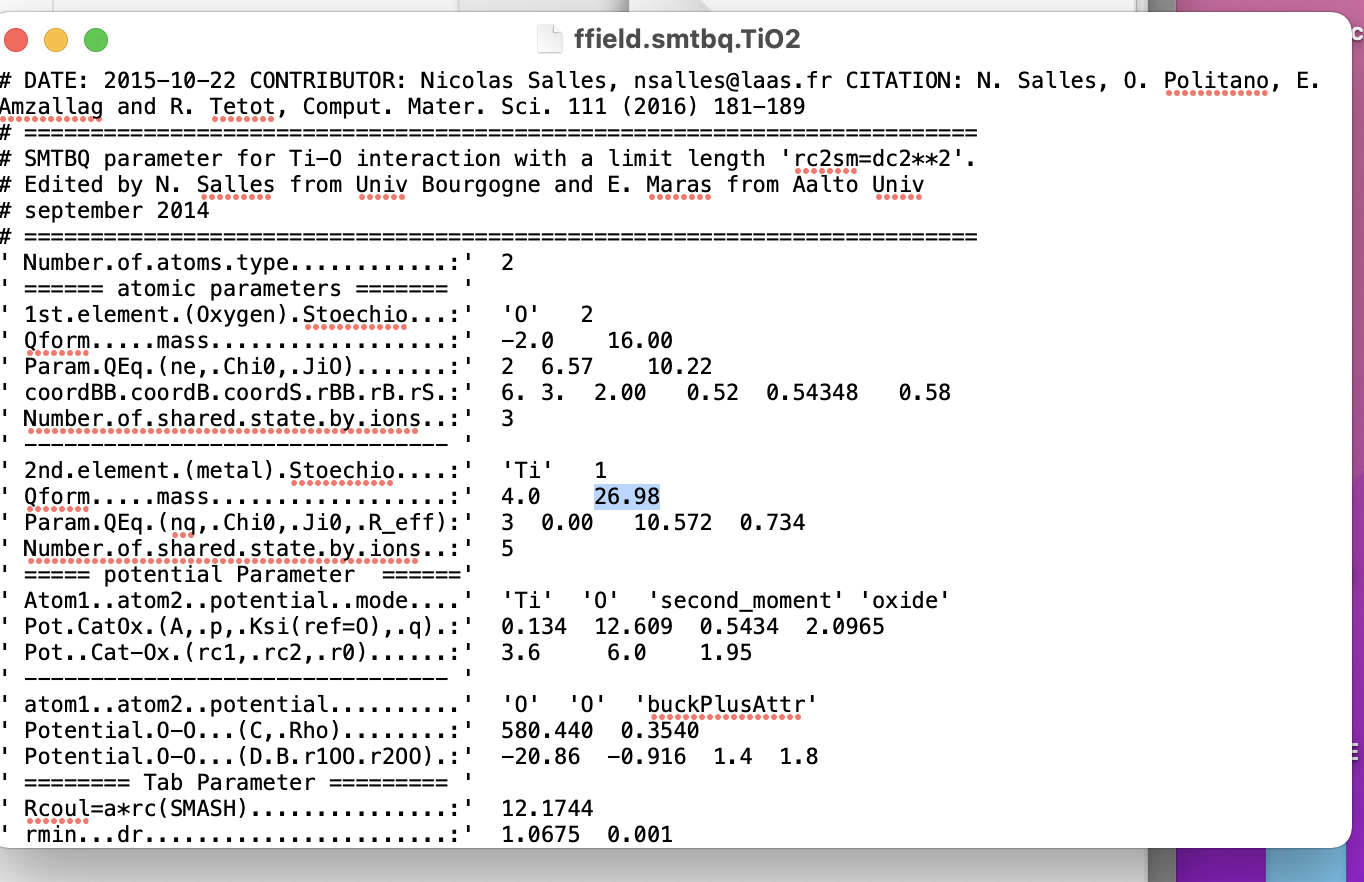
2) I have been trying to find the correct potentials for these three atom types. Especially for Ti-Si and Si-O pairs. Need your help if it is true so far in my script below and what are the best potentials for these pairs?
Attahched is my input script:
with the data file above…
---------- INPUT SCRIPT ---------------------
clear
units metal
dimension 3
boundary p p s
atom_style atomic # Should I change it here?
---------- Create Atoms ---------------------
lattice diamond 5.431
#region box block 0 5 0 5 0 5 units lattice
#create_box 1 box
#create_atoms 1 box
#mass 1 28.0855
#----------------------------------------------
read_data SiSurface.data
mass 2 47.867 #Ti
mass 3 16.000 #O
region surface block 0 55 0 55 33 65 units box
region mid block 0 55 0 55 17 33 units box
region bottom block 0 55 0 55 0 17 units box
group surface region surface
group mid region mid
group bottom region bottom
group Si type 1
group Ti type 2
group O type 3
----HELP!------ Define Interatomic Potential !!! ---------------------
I need Ti-Si and O-Si interaction potentials here, which one?
pair_style hybrid tersoff buck/coul/long 2.5
pair_coeff 1 1 tersoff 1 Si.tersoff Si
I am getting ERROR for this line!
ERROR: Incorrect args for pair coefficients (../pair.cpp:805)
Last command: pair_coeff 1 1 tersoff 1 Si.tersoff Si
pair_coeff 2 2 buck/coul/long 2.5 31120 0.154 5.250 # Ti-Ti
pair_coeff 3 3 buck/coul/long 2.5 11783 0.234 30.220 # O -O
pair_coeff 3 2 buck/coul/long 2.5 16958 0.194 12.590 # O -Ti
kspace_style ewald 1.0e-4
neighbor 1.0 bin
neigh_modify every 1 delay 10 check yes
---------- Define Settings ---------------------
compute eng all pe/atom
compute eatoms all reduce sum c_eng
---------- Run Minimization ---------------------
reset_timestep 0
min_style cg
minimize 1e-12 1e-12 5000 10000
thermo 1000
thermo_style custom step pe lx press temp
#!!!PLEASE NOTE THAT DAMP PARAMETERS ARE IN TIMEUNITs here in PicoSec, it is a good practice
to have damping at every 100 TIMESTEPS so here 0.1Ps=100fs which is 100 timesteps in metal
#units!!!
velocity all create 300 3243434
#fix 1 all npt temp 300 300 0.1 iso 0.0 0.0 1.0
fix 1 surface nve
fix 2 mid nvt temp 300 300 0.1
fix freeze bottom setforce 0.0 0.0 0.0
fix dep Ti deposit 3000 2 1000 342535 region surface vz -1.0 -0.8
fix dep O deposit 3000 3 500 342535 region surface vz -1.0 -0.8
dump 1 all custom 1000 Diamond_dumps/Ti_depon_Si.dumps.* id type x y z
run 100000