Hi,
I want to conduct GCMC simulations to investigate methane adsorption on kerogen by using fix gcmc.
Before that I want to try the simulation of bulk methane using TraPPE force field.
I encountered some problems when calculating the density profile:
-
When I specified Chemical potential (data from NIST database), the density after equilibrium is much higher than expected. Temperature continues increasing, higher than the specified value. Pressure is decreasing but far higher than pressure specified (Several orders of magnitude).
-
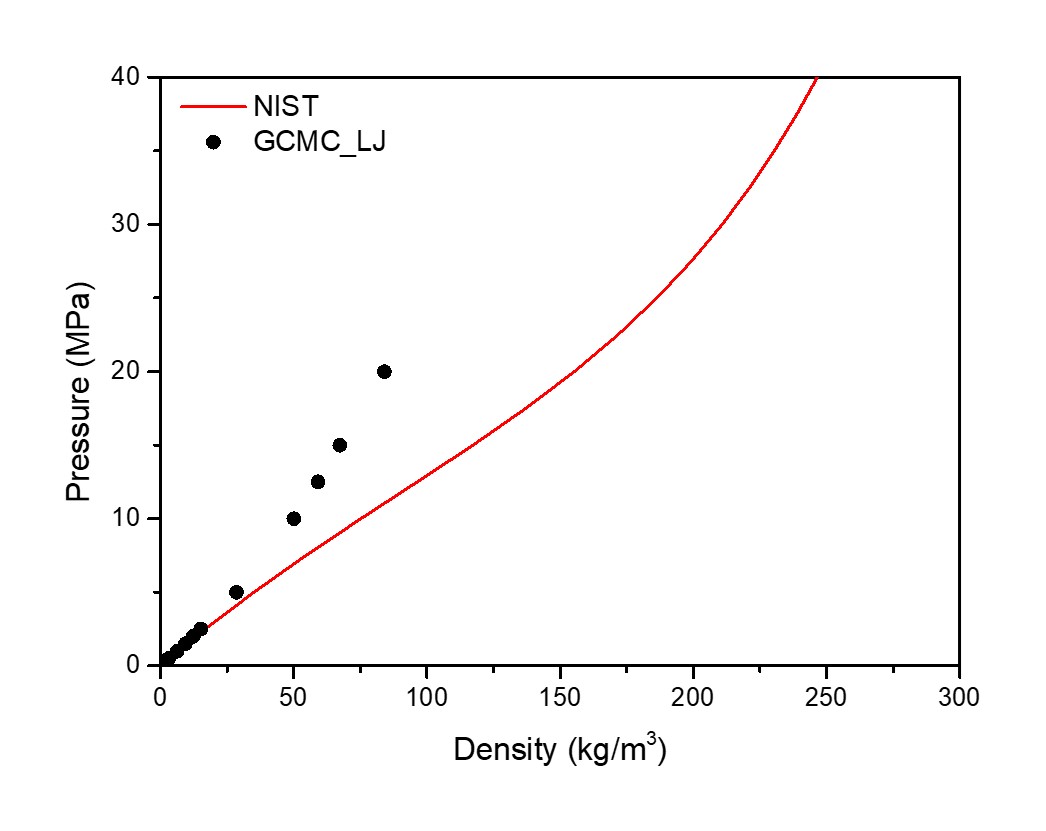
When I specified Pressure and Fugacity coefficient (also from NIST database),
For pressure less than 10 bar, the density profile agrees well with NIST database;
When pressure higher than 10 bar, the density calculated is much lower than expected and the error gets higher with the increase of pressure. Temperature and pressure are both lower than specified. e.g. T specified is 300 K, p specified is 200 bar, fugacity coefficient is 0.75842, density expected is 0.155 g/cm3, while T calculated is around 160 K, p calculated is around 140 bar, density calculated is 0.084 g/cm3. Attached please find the results using Pressue and Fugacity coefficient.
- I guess the problem might be that the temperature was not thermostated at the specified value. So I added the fix_nvt command to the input file. The fix nvt was combined with fix_modify dynamic/dof yes and fix_modify temp as the LAMMPS Manuals says. However, an error was produced as follows: ERROR: Lost atoms: original 10036 current 9730 (…/thermo.cpp:442).
I am not familiar with gcmc at LAMMPS. I have looked through the LAMMPS Mailing List and solved many problems but I haven’t found the answers to the above three problems.
The version of LAMMPS I am using is lammps-17Nov16. I have also tried the version of lammps-11Aug17. But the problem remains unsolved and the speed of calculation slows down and I haven’t found the reason.
Any help would be helpful.
Thank you very much!
Best regards,
Juan
#####################INPUT SCRIPT############################
units real
boundary p p p
atom_style atomic
region mybox block -60.0 60.0 -60.0 60.0 -60.0 60.0
create_box 1 mybox
create_atoms 1 random 10000 3576738 mybox
group mygas type 1
mass 1 16.0425
pair_style lj/cut 10.0
pair_coeff 1 1 0.148 3.730
pair_modify tail yes
velocity mygas create 300.0 4928459 mom yes rot yes dist gaussian
variable chempot equal -1.201599293
variable fugacoeff equal 0.75842
variable pressatm equal 200*0.9869233
#compute mdtemp mygas temp
#compute_modify mdtemp dynamic/dof yes
#fix mdnvt mygas nvt temp 300.0 300.0 100.0
#fix_modify mdnvt temp mdtemp
#fix 2 mygas gcmc 10 1000 1000 1 3456543 300.0 {chempot} 0.1 pressure {pressatm} fugacity_coeff ${fugacoeff}
fix 2 mygas gcmc 10 1000 1000 1 3456543 300.0 ${chempot} 0.1
timestep 1
thermo 500
thermo_style custom step temp press density pe ke etotal
run 200000