I have calculated chemical potentials for a 3 component phase diagram using pymatgen. However, I do not fully understand how chemical potential is calculated. Are temperature corrections taken into account (especially for gas phase)? Is there an attribute I can use to see which structure is being used to calculate the chemical potential?
Please see my code attached and a snippet of the output below. Sample Code
from pymatgen.core.composition import Composition
from pymatgen.core.periodic_table import Element
from pymatgen.ext.matproj import MPRester
from pymatgen.analysis.phase_diagram import PhaseDiagram
#Add your API key here
api_key = None
#Get data for 3 component system
with MPRester(api_key) as m:
entries = m.get_entries_in_chemsys(['Fe', 'Ti', 'O'])
#Generate phase diagram
pd = PhaseDiagram(entries)
#Generate reaction pathways
element_profile = pd.get_element_profile(element=Element('O'), comp=Composition('FeTiO3'))
#Print relevant data
for i, evolution_step in enumerate(element_profile):
print('Reaction: {}'.format(evolution_step['reaction']))
print('Chemical Potential: {}'.format(evolution_step['chempot']))
print('Reaction object fields: {}'.format(evolution_step['reaction'].__dict__.keys()))
print('Components of chemical reaction data:')
for comp in evolution_step['reaction']._all_comp:
print('\t{}'.format(comp.__dict__))
print('-')
Snippet of Output
Reaction: TiFeO3 + 0.25 O2 -> TiO2 + 0.5 Fe2O3
Chemical Potential: -4.93552791875
Reaction object fields: dict_keys([’_input_reactants’, ‘_input_products’, ‘_all_comp’, ‘_els’, ‘_coeffs’])
Components of chemical reaction data:
{‘allow_negative’: False, ‘_natoms’: 5.0, ‘_data’: {Element Fe: 1.0, Element Ti: 1.0, Element O: 3.0}}
{‘allow_negative’: False, ‘_natoms’: 12.0, ‘_data’: {Element Ti: 4.0, Element O: 8.0}}
{‘allow_negative’: False, ‘_natoms’: 40.0, ‘_data’: {Element Fe: 16.0, Element O: 24.0}}
{‘allow_negative’: False, ‘_natoms’: 1.0, ‘_data’: {Element O: 1.0}}
-
I’ll need to double check this, but I believe the chemical potentials printed here are the energy per atom that would be assigned to the O2 species (treated as an independent variable in context with the other entry energies as determined from our simulations). Thus, there isn’t a corresponding structure per se.
If you want a more explicit reference, you can use the energy per atom of the most stable oxygen energy in the MP database (mp-12957, -4.9355 eV/atom). Because of MP’s correction scheme, this value roughly corresponds to a reference chemical potential of O2 gas at STP. You could then estimate the change in chemical potential as a function of T and P using some model (e. g. an equation of state).
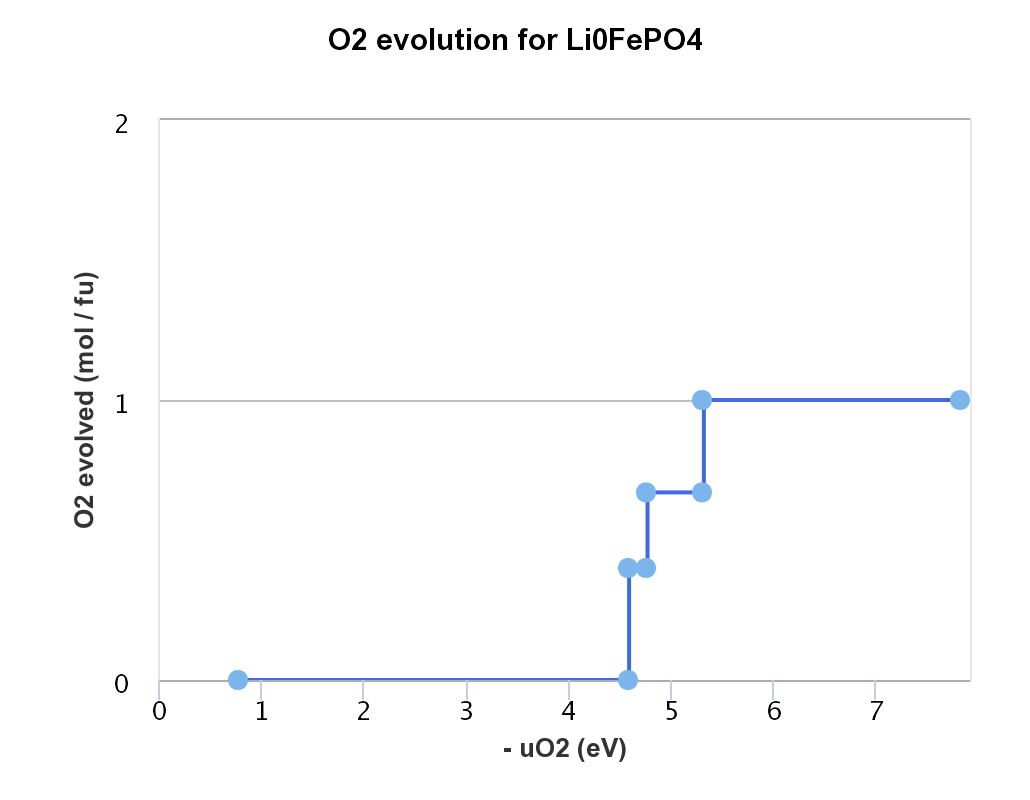
I have a question related to the reference of chemical potential of O2. I tried to replicate the O2 evolution diagram for FePO4 in the battery explorer on the Materials Project website. I can reproduce different ‘steps’ of O2 evolution with the right decomposition reactions but couldn’t figure out how to get the same values of muO2. Here is the code I used:
Code snippet
from pymatgen import MPRester, Composition, Element
from pymatgen.io.vasp import Vasprun
from pymatgen.analysis.phase_diagram import *
from pymatgen.entries.computed_entries import ComputedEntry
from pymatgen.core.composition import Composition
from pymatgen.entries.compatibility import MaterialsProjectCompatibility
ref_entries = [e for e in entries if e.composition.reduced_formula == ‘O2’]
u_ref_0 = min(ref_entries, key=lambda e: e.energy_per_atom).energy_per_atom
print(u_ref_0)
el_profile = pd.get_element_profile(Element(ref_element), entry.composition)
for i, d in enumerate(el_profile):
voltage = d[“chempot”] - u_ref_0
print(“Voltage: 0.5s eV" voltage)
print(d[“reaction”])
print(”")
Here is what I got:
Voltage: 0.0 eV
4 FePO4 -> 4 FePO4
Hi SSeiha,
The battery database is currently out of date so some of the materials in the muO2 plot are missing on the website. If you generated your own muO2 plot and it looks qualitatively similar then I wouldn’t worry about it.
Best,
-Jimmy
Dear Mr.jshen,
Thank for your helping,
Because I am a beginer for Pymatgen, so is this code for jupyter notebook or for other environments?
I usually use code samilar with codes in the bellow picture:
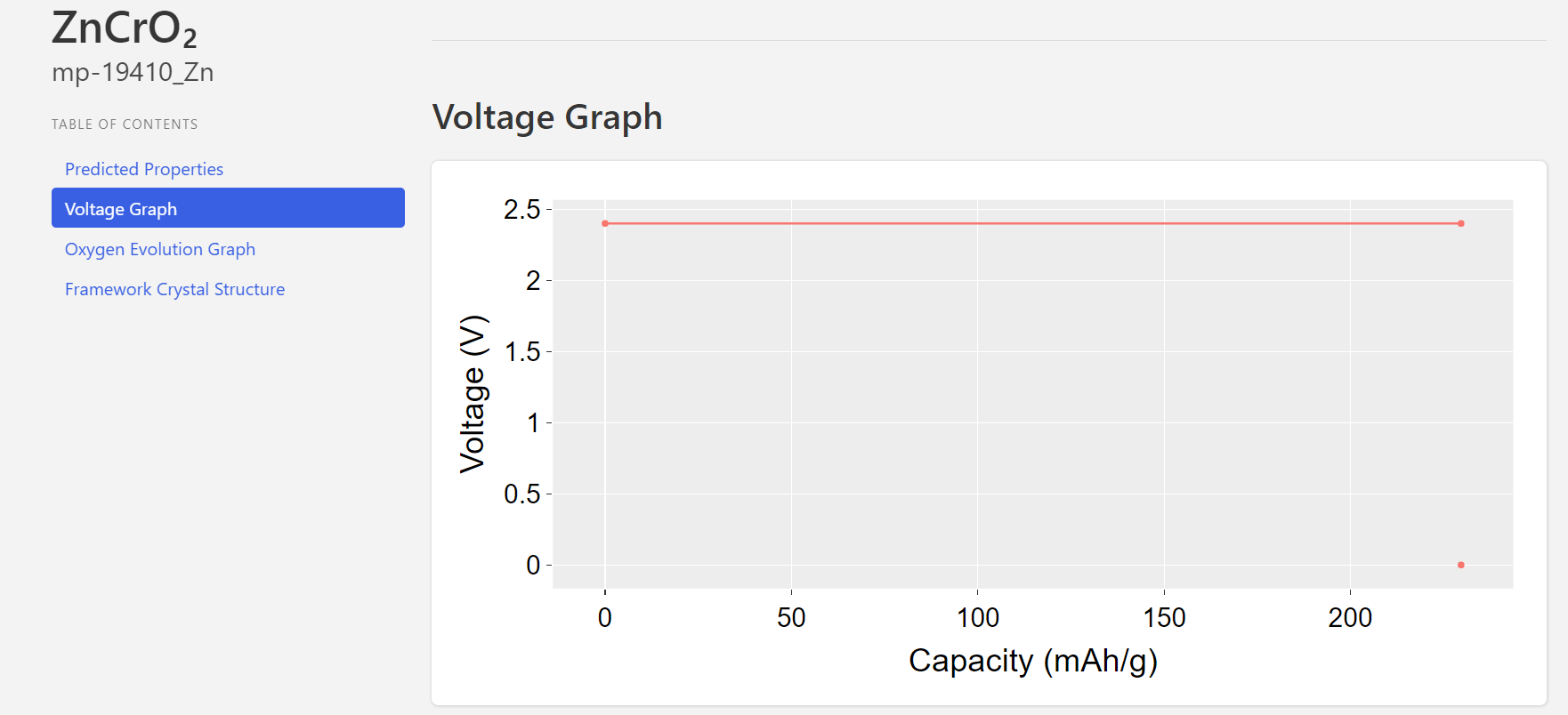
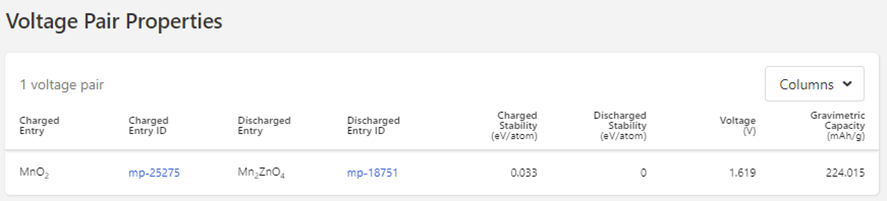
Voltage: 1.520648380000003 V
Mn2ZnO4 → 2 MnO2 + Zn
However, when I am check the voltage graph of ZnMn2O4 (mp-18751) in Material Projects
, it show different value ( 1.619 V):