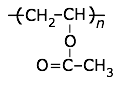The “oplsaa.prm” file does not contain structures. It contains atom properties, force-field parameters, and rules for creating angle, dihedral, and improper interactions for organic molecules.
Determining the atom-types for a molecule requires some care. Perhaps someone can suggest an easier way to do this than what I am doing? (…using 3rd-party software, perhaps? I’m CCing this to Jason Lambert who wrote oplsaa_moltemplate.py)
Anyway, to define a new type of molecule I carefully read through the “atom” and “charge” sections of the “oplsaa.prm” file (available at http://dasher.wustl.edu/tinker/distribution/params/oplsaa.prm).
You can usually figure out the type of each atom by looking at
-
its description (in quotes, 5th column of “atom” section),
-
its atom-type-name (4th column of “atom” section)
-
its mass (6th column of “atom” section), and
-
its charge: (3rd column of the “charge” section)
For example, there are two atoms named “Ammonia NH3”, one with mass 14.007, and the other with mass 1.008:
atom 78 44 NT “Ammonia NH3” 7 14.007 3
atom 79 45 H “Ammonia NH3” 1 1.008 1
Currently, the first 56 atom types are united atoms “(UA)” (a form of coarse-graining) and should probably not be used in an all-atom simulation.
Unfortunately, different versions of the “oplsaa.prm” file use different atom type numbers. So you have to re-do this every time you download a newer version of the “oplsaa.prm” file. (The descriptions and names are usually the same.)
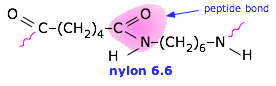
I attempted to pick atom types for the two polymers in your example below. However I was not very careful, and my knowledge of organic chemistry is rather poor. There are probably better people to give you advice than me.
If you guess incorrectly, then hopefully LAMMPS may complain that your system is not neutral. This is a good way to catch your mistakes:
http://lammps.sandia.gov/threads/msg50912.html
http://sourceforge.net/p/lammps/mailman/message/33238059/
See below for my suggestions for your molecule:
P.S.
— Official documentation:—
If it helps, the “oplsaa.prm” file is distributed with TINKER and maintained by the TINKER developers. Read about it here:
http://dasher.wustl.edu/tinker/
http://dasher.wustl.edu/tinker/downloads/guide.pdf
… and if I understand correctly, the contents of the “oplsaa.prm” file are extracted from the BOSS documentation located here:
http://zarbi.chem.yale.edu/software.html