I am currently working on a topic “Mechanical Characteristics of Copper-Coated SWCNTs in Al-Cu-Mg Metal Matrix Composites”. At the moment, I am encountering some challenges with the code related to my study.
Problem :
When I am running my code, the CNT inside the coating is getting distorted. I have taken dump after using NPT ensemble and the error is occuring. I am not able to understand the proper reason why this is happening.
This my model:
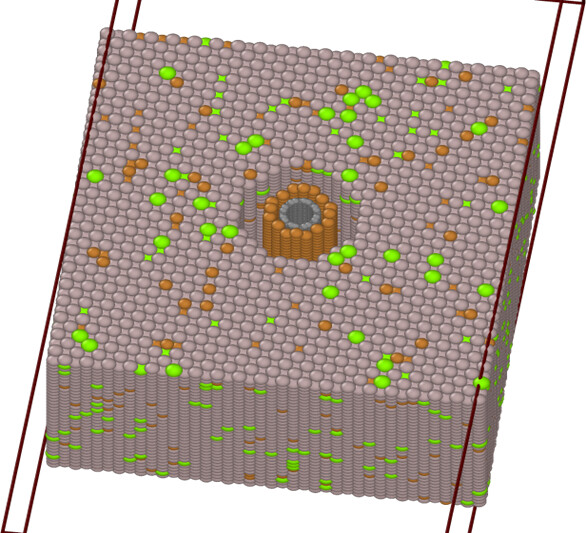
This my model after running my code:

My Code here:
units metal
variable T equal 300
variable dt equal 0.0001
dimension 3
boundary p p p
atom_style atomic
read_data final.lmp
Assign atom types (1=Al, 2=Cu, 3=Fe, 4=C)
mass 1 26.98 # Al
mass 2 63.546 # Cu
mass 3 23.305 # Mg
mass 4 12.01 # C
Define interatomic potentials using hybrid
pair_style hybrid airebo 10.2 eam/alloy lj/cut 9.5
AIREBO potential for C-C interactions
pair_coeff * * airebo CH.airebo NULL NULL NULL C
LJ potential for Al-C, Cu-C & Mg-C
pair_coeff 1 4 lj/cut 0.03438 3.01
pair_coeff 2 4 lj/cut 0.02578 3.0825
pair_coeff 3 4 lj/cut 0.0027 3.5015
EAM potential for Al-Cu-Mg interactions
pair_coeff * * eam/alloy CuAgAuNiPdPtAlPbFeMoTaWMgCoTiZr_Zhou04.eam.alloy Al Cu Fe NULL
Define simulation parameters
neighbor 2.0 bin
neigh_modify delay 0 every 1 check yes
###------------------energy #minimization-----------------------------
min_style cg
minimize 1.0e-6 1.0e-6 10000 10000 # Energy tolerance = 1.0e-6, Force tolerance = 1.0eV/Å
Step 2: Thermal Equilibration under NPT
Nose-Hoover thermostat and Parrinello-Rahman barostat-
velocity all create 300.0 5812775 mom yes rot yes dist gaussian # Initialize at 300 K
fix 1 all npt temp 300.0 300.0 (100.0*dt) iso 0.0 0.0 (100.0*dt) # NPT ensemble, 0 bar pressure
timestep 0.0001
thermo 100
dump 1 all custom 100 dump_n1.lammpstrj id type x y z
run 400000
Step 3: Uniaxial Tensile Deformation in z-direction (NVT Ensemble)
unfix 1
fix 2 all nvt temp 300.0 300.0 $(100.0*dt) # NVT ensemble at 300 K
fix 3 all deform 1 z erate 1.0e-4 remap x # Apply strain rate of 10^-4 /ps in z-direction
timestep 0.0005 # Time step of 0.5 fs for deformation
I would greatly appreciate any advice or resources you could offer.
Step 4: Output data (thermo and dump files)
thermo 100
thermo_style custom step temp press pe ke lx ly lz
dump 2 all custom 100 dump_n2.lammpstrj id type x y z
run 8000000