Hi,
I am facing an issue, and this is my very last one for the simulations I am working on:
“ERROR: Out of range atoms - cannot compute PPPM (…/pppm_tip4p.cpp:107)”
I am certain that the cause of this is that I am missing or am using incorrect pair coefficients, to model a sheet of hydroxylated/hydrogenated porous graphene with airebo and water with the LJ/tip4p model.
I have 11 atom types.
-
Graphene: C, zCo, zO, zHo, zCh, zHc (“z” indicates atom on pore edge, capital letter is type; first 3 after C are from -COH and last 2 are from -CH)
-
Saltwater: H, O, CL, NA
-
piston (carbon-based)
pair_style hybrid lj/cut/tip4p/long {Otype} {Htype} {waterbond} {waterangle} 0.1546 13.0 airebo 3.0 0 1
C CL H O piston NA zCo zCh zO zHc zHo
pair_coeff * * airebo CH.airebo_real C NULL NULL NULL NULL NULL NULL C NULL H NULL
pair_coeff {Ctype} {CLtype} lj/cut/tip4p/long 0.0317022 4.2821 # C-CL
pair_coeff {Ctype} {Htype} lj/cut/tip4p/long 0 0 # C-H
pair_coeff {Ctype} {Otype} lj/cut/tip4p/long 0.118238 3.28203 # C-O
…
The rest are attached in the “pair_coeffs” file. Can somebody please explain or direct to me to which pair_coeffs I need to be using or that I may be missing, and why? These values are calculated from a table in Cohen-Tanugi’s thesis appendix.
This is urgent, and I appreciate any help or advice, as soon as possible.
Thanks,
Apoorv Khandelwal
pair_coeffs.txt (4.6 KB)
log.lammps (10.9 KB)
single.in (10 KB)
single_system.dat (424 KB)
For some reason, I think the body of my mail has failed to appear:
Hi,
I am facing an issue, and this is my very last one for the simulations I am working on:
“ERROR: Out of range atoms - cannot compute PPPM (…/pppm_tip4p.cpp:107)”
I am certain that the cause of this is that I am missing or am using incorrect pair coefficients, to model a sheet of hydroxylated/hydrogenated porous graphene with airebo and water with the LJ/tip4p model.
I have 11 atom types.
-
Graphene: C, zCo, zO, zHo, zCh, zHc (“z” indicates atom on pore edge, capital letter is type; first 3 after C are from -COH and last 2 are from -CH)
-
Saltwater: H, O, CL, NA
-
piston (carbon-based)
pair_style hybrid lj/cut/tip4p/long {Otype} {Htype} {waterbond} {waterangle} 0.1546 13.0 airebo 3.0 0 1
C CL H O piston NA zCo zCh zO zHc zHo
pair_coeff * * airebo CH.airebo_real C NULL NULL NULL NULL NULL NULL C NULL H NULL
pair_coeff {Ctype} {CLtype} lj/cut/tip4p/long 0.0317022 4.2821 # C-CL
pair_coeff {Ctype} {Htype} lj/cut/tip4p/long 0 0 # C-H
pair_coeff {Ctype} {Otype} lj/cut/tip4p/long 0.118238 3.28203 # C-O
…
The rest are attached in the “pair_coeffs” file. Can somebody please explain or direct to me to which pair_coeffs I need to be using or that I may be missing, and why? These values are calculated from a table in Cohen-Tanugi’s thesis appendix.
This is urgent, and I appreciate any help or advice, as soon as possible.
Thanks,
Apoorv Khandelwal
Here are the attachments. Please disregard the other thread.
log.lammps (10.9 KB)
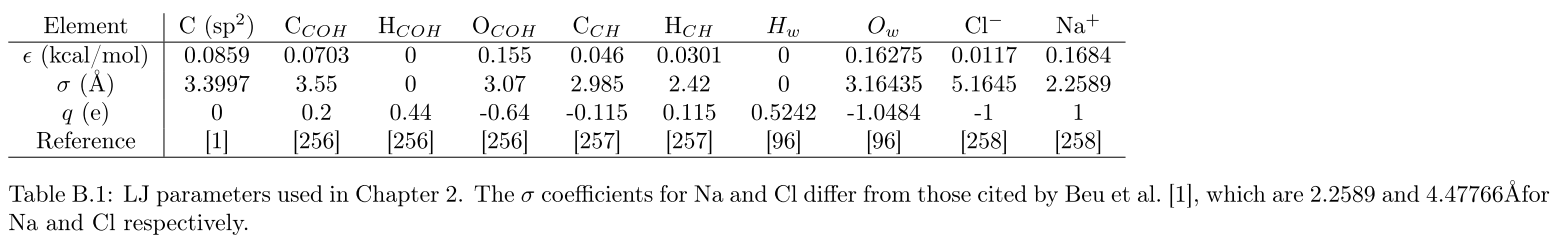
single.in (10 KB)
single_system.dat (424 KB)
pair_coeffs.txt (4.6 KB)
I think your potential energy is way too high. This will in turn lead to huge forces and hence lost atoms.
Did you visualise your initial configuration? Is there an unrealistic amount of overlap between particles?
I have taken a look at the initial configuration through VMD, there does not seem to be any overlap in the LAMMPS data file. I was successfully able to model this same simulation with pristine graphene, but as soon as I introduce pores (-COH), I am forced to change pair coefficients and the simulation fails.
How can I adjust my potential energy properly? All of my values are calculated from the thesis appendix. Am I correct in wanting AIREBO potentials between the carbons and hydrogens in graphene, including -COH and -CH of pores, with LJ/Coulumbic interactions between all other pairs? What about the oxygen in -COH?
I really have no idea, I have zero experience with these force fields. I do know that you forgot to include some potential files, on my computer the run errors on “Cannot open AIREBO potential file CH.airebo_real”.
CH_airebo.real file is attached, sorry forgot to include it initially.
CH.airebo (770 KB)
I really have no idea, I have zero experience with these force fields. I do
know that you forgot to include some potential files, on my computer the run
errors on "Cannot open AIREBO potential file CH.airebo_real".
let me repeat what steve already mentioned: using a CH.airebo
potential file converted to real units is a *very* bad idea.
validating such a file is a major undertaking and something that only
*very* experienced people should do, or at the very least experienced
people under the close supervision of a very experienced person. it is
*much* easier to the other parameters from kcal/mol to eV and similar.
debugging a complex input for a complex system is not something that
people can easily do by just glancing at them.
often mistakes only show up after statistical analysis of the results.
axel.
I understand, I am using a CH.airebo_real file, referenced to me by Cohen-Tanugi, as used in very similar simulations in his thesis.
The source was: https://sites.google.com/site/mechapple1501/pages/CH.airebo_real?attredirects=0&d=1
If you know more, would you be able to tell me more about using pair_coeffs and the airebo or lj/tip4p force fields, so I may debug more myself? Which atom types should I apply these to or is there another field I may need?
I understand, I am using a CH.airebo_real file, referenced to me by
Cohen-Tanugi, as used in very similar simulations in his thesis.
The source was:
https://sites.google.com/site/mechapple1501/pages/CH.airebo_real?attredirects=0&d=1
If you know more, would you be able to tell me more about using pair_coeffs
and the airebo or lj/tip4p force fields, so I may debug more myself? Which
atom types should I apply these to or is there another field I may need?
you have a *very* convoluted and complex system. even for an
experienced person, it will take a considerable amount of time and
effort to break it down in pieces and step by step check whether
everything is working correctly or not.
everything you would need to know about force field coefficients and
related is readily available in text books on MD. but as i mentioned
before, this is not something you can pick up in a jiffy.
axel.
