Hi all,
I’m trying to calculate the potential of mean force for a methanol molecule solvated in
a slab of water. I am using COLVARS and the abf method. The simulation runs fine and
the molecule moves along the target centers.
-
What are the optimal values for lowerwallConstant, upperwallConstant, forceConstant?.
-
Does the PMF obtained need to be rescaled depending on the value input for the forceConstant ?, e.g. if the forceConstant=200 then divide the final PMF values by 200?.
Thanks a lot and regards,
Adriano
Hello Adriano, I’m not sure why you mention target centers and ABF, which doesn’t use any restraint centers, but only a history-dependent force.
If you are using restraints on top of ABF, please be aware that these restraints will be accounted for in the resulting PMF for a recent enough version:
http://colvars.github.io/README-totalforce.html
The optimal value of force constants is highly system dependent, and you are best off consulting the literature and/or running short test simulations initialized near the boundaries that you defined.
Any PMF written by the Colvars module has the free energy in the units that you are using in the LAMMPS simulation. There is no rescaling needed unless you wish to change that unit.
Giacomo
Hi Giamoco,
Thanks for your email. The ‘targetcenters’ comes from the ‘harmonic {}’ section in the colvars file.
If I don’t add it then LAMMPS produces a warning message when it begins to run the COLVARS module:
colvars: Warning: lowerWallConstant and lowerWall are deprecated, please define a harmonicWalls bias instead.
This is the COLVARS file:
colvarsTrajFrequency 1000 # output every 10 steps
colvarsRestartFrequency 1000
colvar {
name dist
width 1 # in Armstrongs
lowerboundary 3.0
upperboundary 25.0
lowerwallConstant 100000
upperwallConstant 100000
outputValue
outputAppliedForce
distanceZ {
main { atomNumbers 161 162 163 164 165 166 167 168 169 }
ref { atomNumbersRange 1-160 }
}
}
harmonic {
name abf_pmf
colvars dist
forceConstant 19.987 # energy units/{A}^2
centers 3
targetCenters 25
targetNumSteps 161000000 # numbers of steps to go from d2-d2
}
abf {
name abf
colvars dist
fullSamples 500
historyFreq 1000
}
Hi Adriano, please read the deprecation warning carefully: it recommends defining a harmonicWalls, not harmonic, and instead of the legacy keyword lower/upperWall, not in addition.
Also, why are you putting a moving harmonic restraint (i.e. a SMD protocol) on top of the ABF calculation? You cannot expect a meaningful PMF to come from either one if they are both applying a time-dependent force.
You should probably decide in advance what PMF calculation method you want to use and write the input accordingly. Do not add keywords only to work around warnings or errors.
Giacomo
Hi Giacomo,
Thanks, I have fixed that issue and defined a harmonicwalls. Please see the COLVARS file below.
I ran a long simulation for 160 ns and the PMF has a typical shape. The minima at the solid/liquid
interface I get is -45 kcal mol-1 (LAMMPS REAL UNITS) if measured from the bulk, which I consider
the zero energy level. This value seems to high, I was expecting to get around (-5,-10) kcal mol-1.
COLVARS 1 solvated ETOH molecule adsorbed on graphene.
colvarsTrajFrequency 1000 # output every 10 steps
colvarsRestartFrequency 1000
colvar {
name dist
width 1.0 # in armstrongs
lowerboundary 3.0
upperboundary 25.0
outputValue
outputAppliedForce
distanceZ {
main { atomNumbers 161 162 163 164 165 166 167 168 169 }
ref { atomNumbersRange 1-160 }
}
}
harmonicWalls {
name abf_pmf
colvars dist
lowerWalls 3
upperWalls 25
forceConstant 19.987 #energy units (kca/mol)/{A}^2
}
abf {
name abf
colvars dist
fullSamples 500
historyFreq 1000
}
Thanks,
Adriano
Hi Adriano, how does your system look like?
For a simple 1D PMF along a Cartesian axis, it is possible to use internal forces as the free-energy estimator (in Colvars, this is done by ABF, or by metadynamics and/or harmonic restraints with the writeTIPMF keyword). It is usually hard to beat the internal-forces estimator, and 160 ns is a decent simulation time.
I would look at the trajectory to make sure that the atom selections are OK, and that they select the groups you want throughout. On Linux or Mac builds of VMD, Colvars is pre-built in:
https://colvars.github.io/colvars-refman-vmd/colvars-refman-vmd.html#sec:cv_command_example
Beware of possible wrapping/unwrapping differences between the dump’ed trajectory that VMD will read and the runtime coordinates in LAMMPS (which handles PBCs well through image flags):
Another step in making sure that your setup works well is using it on an octanol/water interface, so that you can compare to a standard measurement. For methanol (logP = -0.7), you should be seeing a very small free-energy difference, thus making a stringent test.
Giacomo
Hi Giacomo,
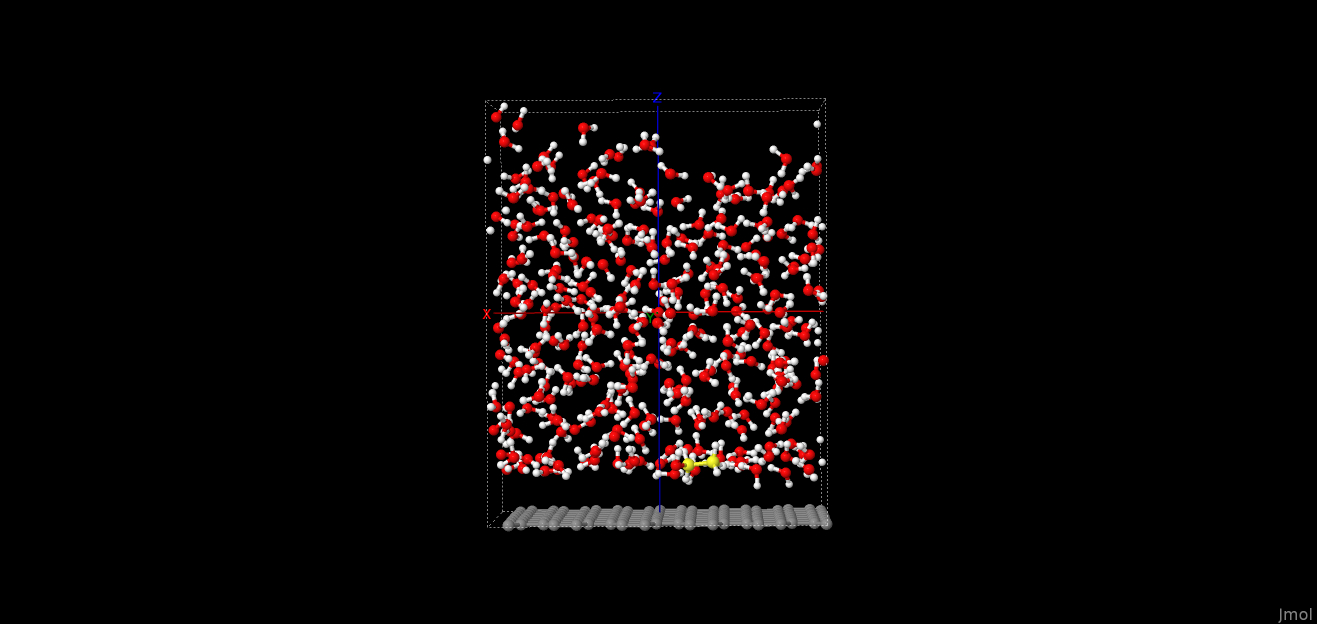
Please see attached a picture of the unit cell. The ethanol molecule is in yellow colour, solvated in water
next to a graphene sheet. It is all in PBC and the cell is enlarged in the z axis.
I am sure the groups are defined properly. This is from the output:
160 atoms in group solute
9 atoms in group ethanol
846 atoms in group solvent
This is in the LAMMPS input file to call the COLVARS module
fix abf all colvars ETOH_216.colvars tstat 3
run 161000000
unfix abf
Thanks,
Adriano
Thanks, in that case I would definitely look at whether the model is physically representative of a bulk water/graphene interface. The water phase needs to be at the correct density (is it?), and it is much preferable to run in the NPT ensemble, so that the volume changes due to adsorption of the molecule will not impact the free energy difference.
Keep in mind that the above are just guesses: otherwise, as far as I can tell the Colvars input looks syntactically correct to me.
Giacomo
Hi Giacomo,
The density is around 0.9-1.0 g/cc and after running the same simulation in the NPT ensemble I pretty much get the same
result. I am trying now to increase the unit cell size but at least I have the right Colvars settings.
Thanks!,
Adriano