Hi,
I intend to do a PMF analysis using ColVars (lammps/15May15). I have read ColVars manual, but one part is not clear enough for me.
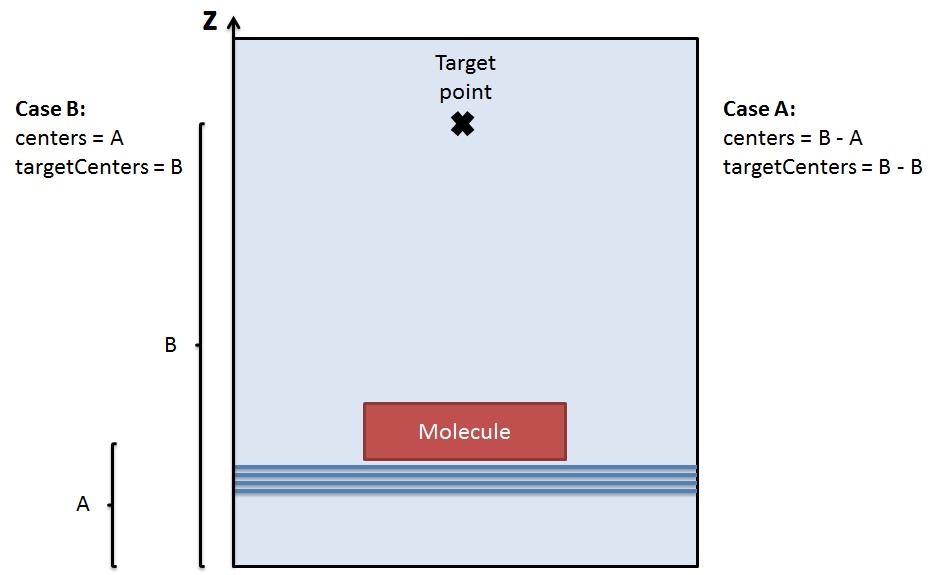
Regarding the centers and targetCenters, which case is correct (see figure)?
I have tried different cases, but don’t see a certain behavior; The object either does not desorb from the surface, or it jumps from the surface and then smoothly moves to the target point.
Thanks so much.
ColVars input:
colvarsTrajFrequency 1
colvarsRestartFrequency 1000
colvar {
name Des
width 1
lowerBoundary 0.0
upperBoundary 18.0
force constants
lowerWallConstant 5000
upperWallConstant 5000
hardLowerBoundary on
outputs
outputAppliedForce on
#distanceZ colvar
distanceZ {
main {
atomNumbersRange 1-18 #ring
}
ref {
dummyAtom (9.97907, 10.0362, 1.97266) #ref
}
axis (0,0,1)
}
}
harmonic {
name Desorb-PMF
colvars Des
centers 16.02734 #go from 1.97266 (18 - 1.97266 = 16.02734) forceConstant 100.0 # constant force targetCenters 0.0 #to 18
targetNumSteps 2000
outputEnergy on
outputCenters on
outputAccumulatedWork on
}