@William_Moriarty , I’m happy for us to keep chatting here if you need occasional help, because documenting sources of inconsistency between different MD engines is important for reproducibility.
With different integrators you should not expect identical trajectories. You should be able to calculate the bond forces from the configurations and confirm that those are consistent.
I really appriciate this! I am sorry that I could not respond sooner. I got occupied by other things that needed my immidiate attention.
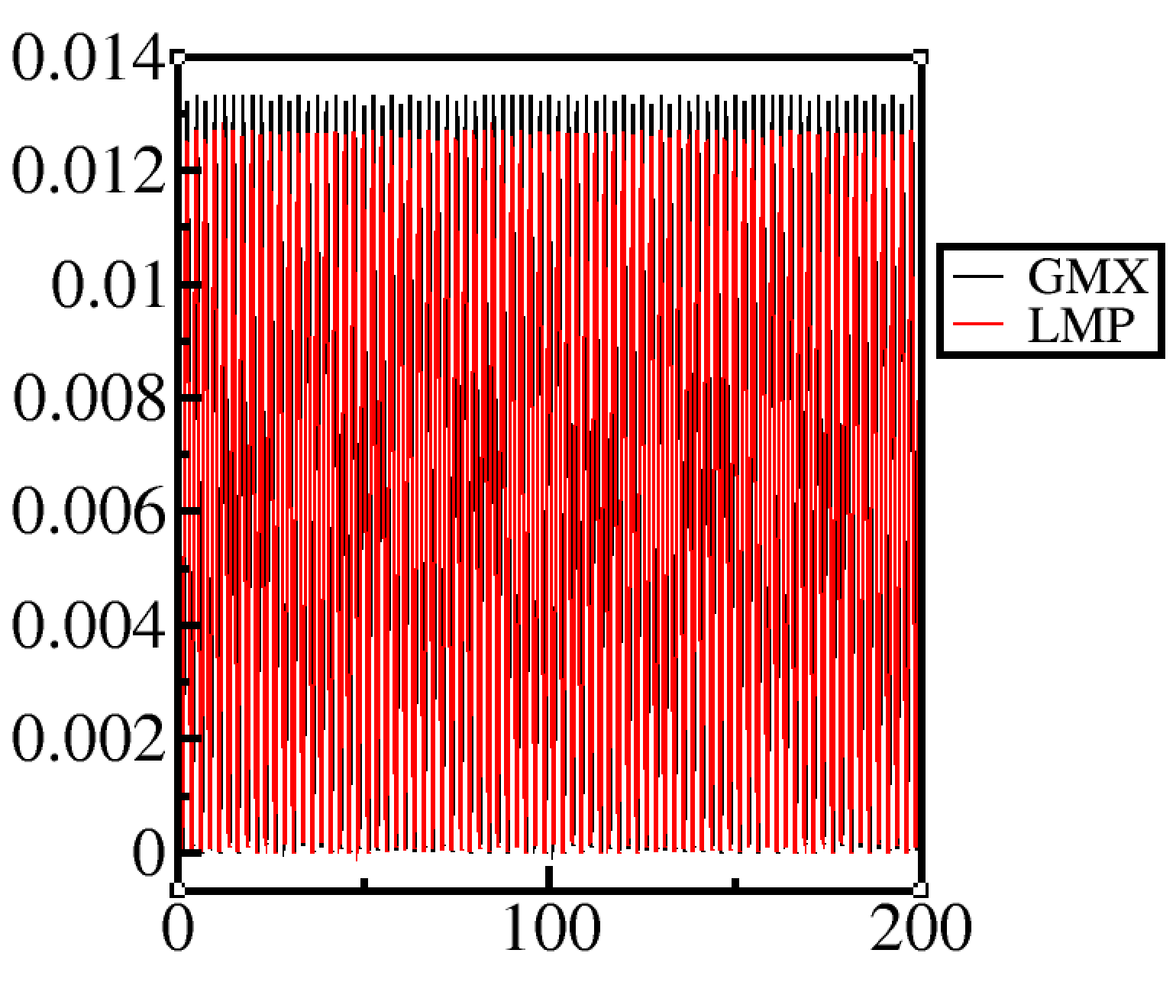
Anyhow, I look at the bond energy and the way I compute is I take k*(bond_length_in_force_field - instantaneous_bond_length)^2 for lammps and 0.5*k*(bond_length_in_force_field - instantaneous_bond_length)^2. The GROMACS energies seem to be a little higher, I dont know if this is ‘comparable’ or not.
(Gromacs does not have the 0.5 baked in the spring constant)
Check that you are using distance outputs to the same precision from both packages (a GROMACS distance output of 1.0 in nm is accurate to the nearest angstrom, while a LAMMPS distance output of 1.0 in angstroms is accurate to the nearest tenth of an angstrom).
Also compare your analytically calculated bond energy to the program-output bond energy.
Finally, for full comparison’s sake, you should ensure both programs are using similar-sized overall simulation boxes and the same boundary periodicities.
Most importantly: it is not meaningful to compare energies against time between the two trajectories. It is very doubtful that GROMACS and LAMMPS were using the same integrators, since their defaults are different. Therefore, you have to compare the energies against configuration – that is, either:
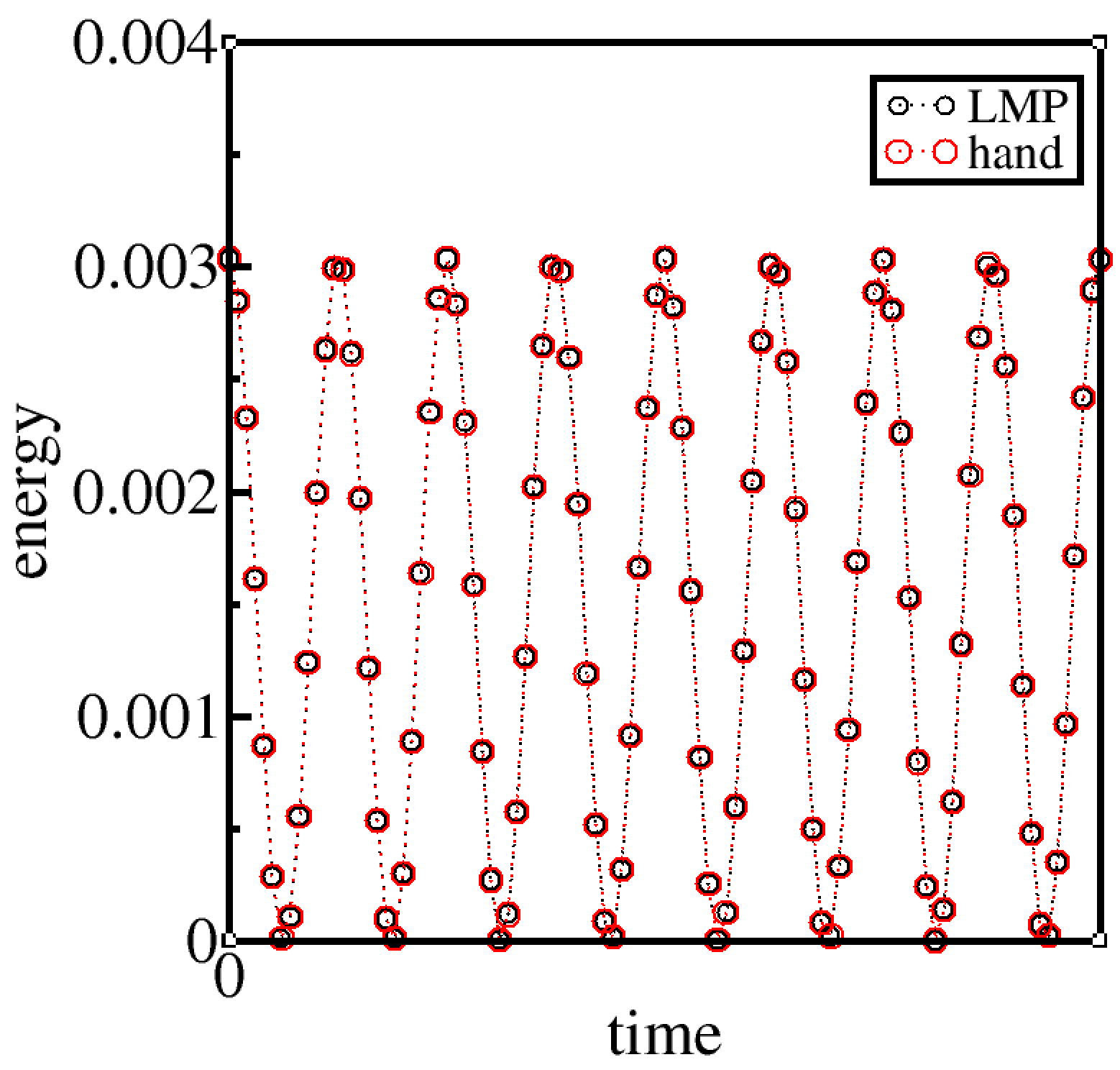
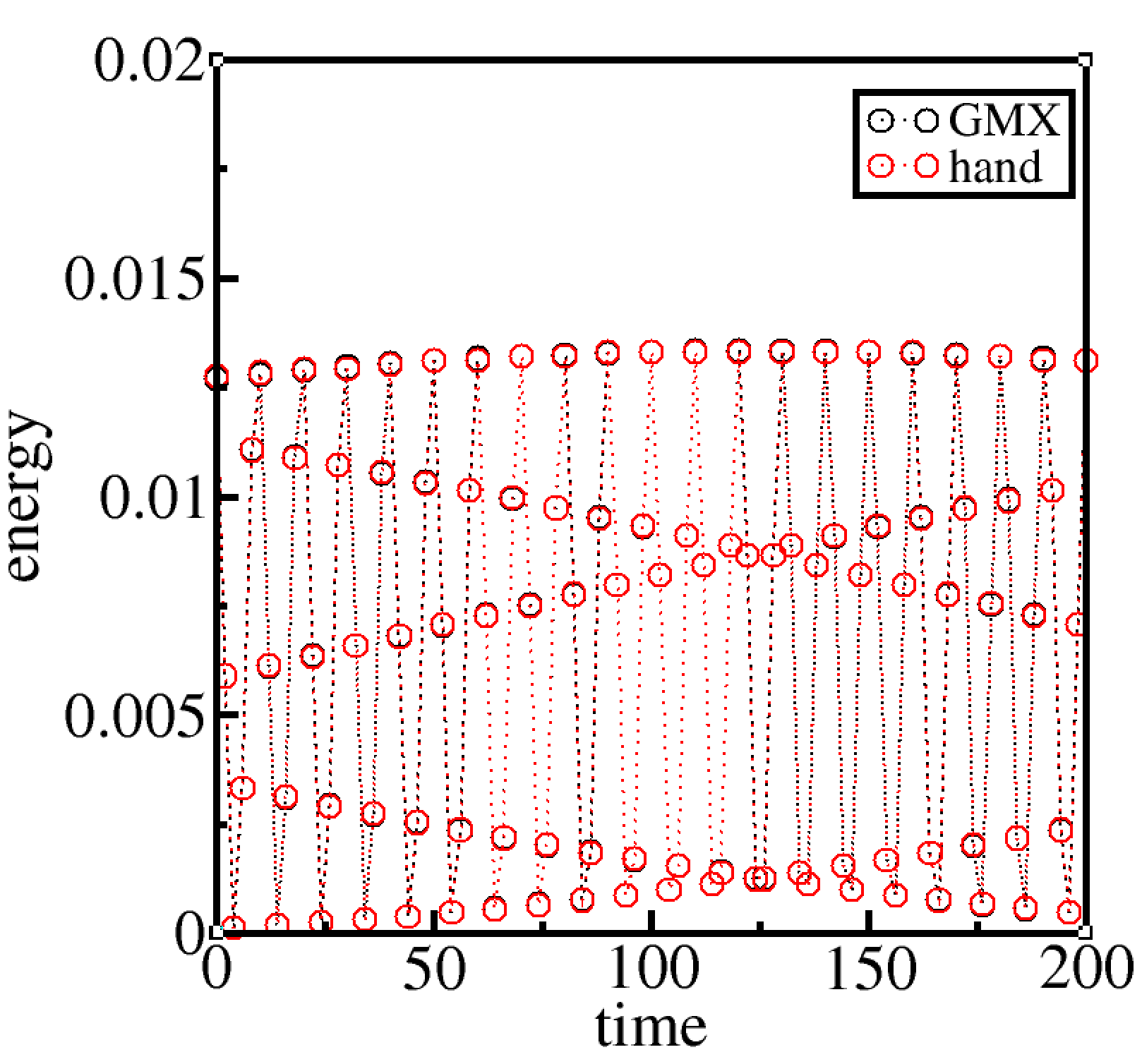
calculate the energies by hand from each configuration and compare them against program output energies, or
calculate the forces by hand from each configuration and compare them against program output forces.
I can’t really do this for GROMACS as gromacs does not output bond energies. So I set epsilon to zero to get the potential energy equal to the bond energy as non-bonded energy is now zero. With epsilon set to zero, the bond energy I compute by hand matches exactly the potential energy output-ed by GROMACS.
…? If GROMACS calculates energies and includes them in thermodynamic output, for forces that it does not include in its dynamics, then that’s not mathematically consistent.
I’d appreciate if you could attach your GROMACS inputs for the dumbbell here, this is pretty scary and I’d like to investigate myself further.
In any case, you now know what to do for the angle and dihedral contributions.
A significant shortcut that may help you: if you can output the GROMACS trajectory and GROMACS forces, you can calculate LAMMPS forces for the GROMACS trajectory using rerun to read the GROMACS trajectory into LAMMPS (last I remember, LAMMPS can read .xtc trajectories with the MOLFILE plugin).
That way you can proceed with direct force-to-force comparisons on the same trajectory.