Hello all,
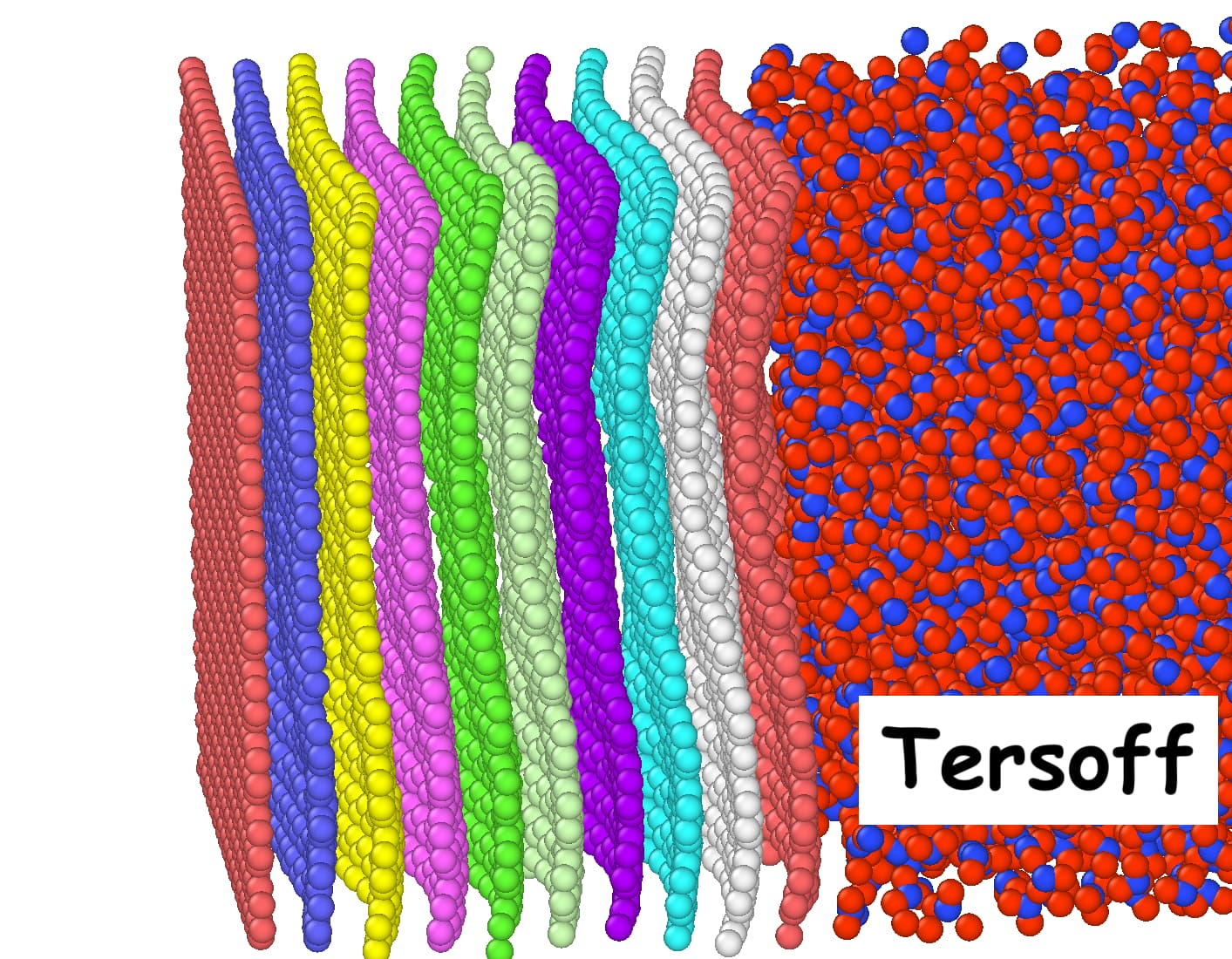
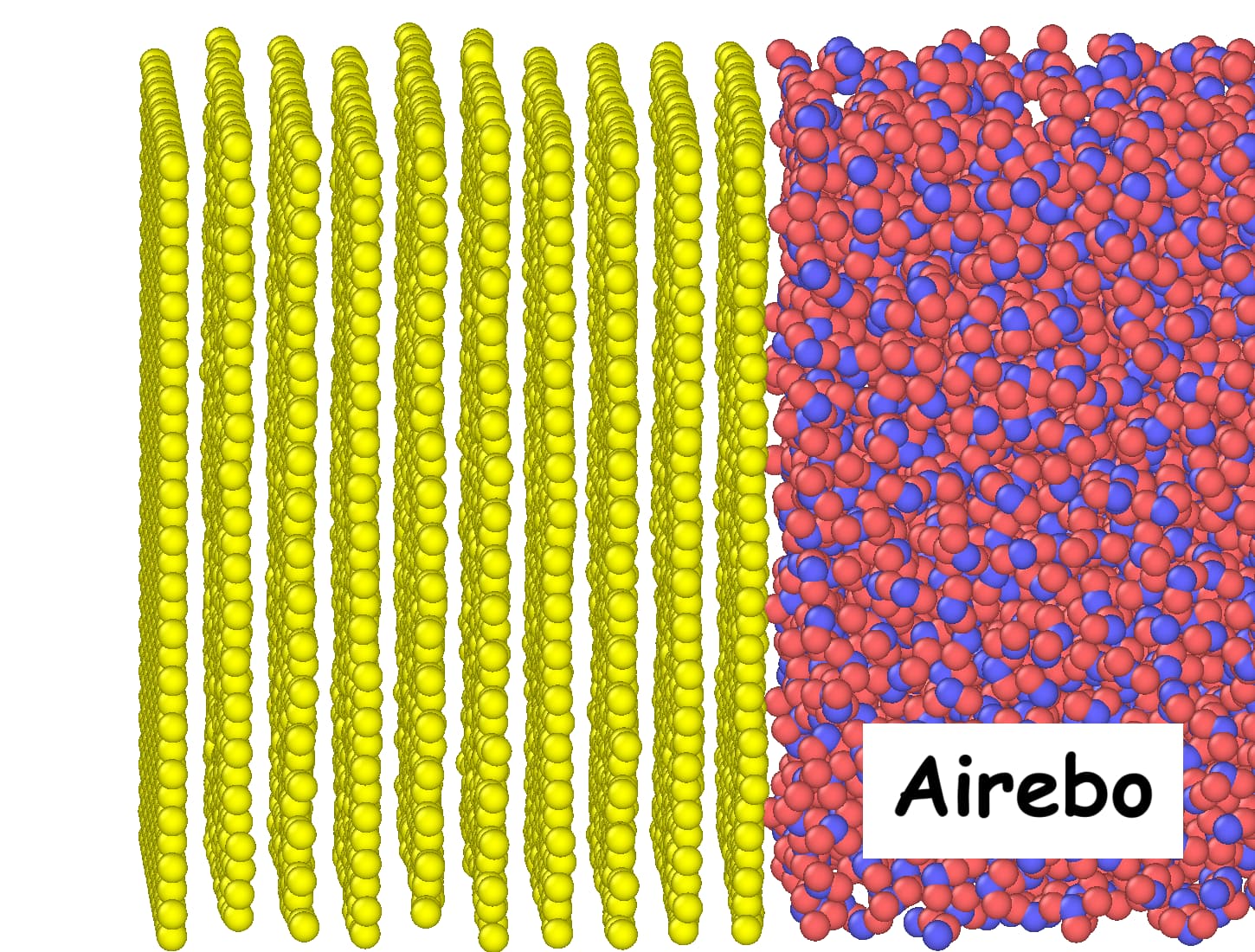
I have implemented the AIREBO potential in my simulations and obtained good agreement with a reference paper regarding Kapitza resistance and heat flux. However, **I’m facing issues when switching to the Tersoff potential: graphene layers deform significantly (bend or warp) during the simulation.Snapshots taken toward the end of both the AIREBO and Tersoff simulations are attached for visual comparison.
The simulation setup follows the LAMMPS manual, relevant discussions on this forum, and Professor Axel Kohlmeyer’s explanations.
Simulation setup:
10 graphene layers on each side, ~4 nm water in between.
Each graphene layer is defined as a separate atom type (only in the Tersoff simulation).
Hot and cold thermostats are applied to the outermost graphene layers to induce a heat flux for Kapitza resistance calculation.
The outermost graphene layers on the left and right sides are fixed in place using fix setforce.
Potential definitions in Tersoff simulation (example for 2 layers):
Blockquote
pair_style hybrid tersoff tersoff lj/cut 10.2 lj/cut/coul/long 12.0
Carbon intralayer
pair_coeff * * tersoff 1 SiC_1994.tersoff NULL NULL C NULL
pair_coeff * * tersoff 2 SiC_1994.tersoff NULL NULL NULL C
Carbon interlayer
pair_coeff 3 4 lj/cut 0.00239721633 3.35
Water-water
pair_coeff 1 1 lj/cut/coul/long 0.0 0.0
pair_coeff 1 2 lj/cut/coul/long 0.0 0.0
pair_coeff 2 2 lj/cut/coul/long 0.00673977503 3.165
Carbon–H
pair_coeff 1 3 lj/cut/coul/long 0.0 0.0
pair_coeff 1 4 lj/cut/coul/long 0.0 0.0
Carbon–O
pair_coeff 2 3 lj/cut/coul/long 0.0040627276 3.190
pair_coeff 2 4 lj/cut/coul/long 0.0040627276 3.190
Blockquote
This pattern is extended accordingly for each of the 20 graphene layers in the full simulation.
To prevent deformation of the graphene sheets, I have separately tested both fix spring and fix spring/self. The reference paper suggests fixing the center of mass (COM) of each graphene layer, so I used fix spring for this purpose. However, it didn’t prevent warping.
Alternatively, I tried fix spring/self, which ties each atom to its initial position. This reduced deformation and matched AIREBO’s behavior more closely. But since this approach constrains individual atoms rather than the COM, it seems unphysical and also alters the heat flux depending on the spring constant.
Also, in the Tersoff simulation without using fix spring/self, the Kapitza resistance is about 13% lower than the expected (reference) value.
Is this kind of deformation expected behavior from the Tersoff potential itself?
If not, could it be due to an issue in the way Tersoff interactions are defined in my input script?
If the Tersoff part is correctly implemented, is the use of fix spring/self a recommended workaround in this context?
What would be the best way to make the behavior of graphene in the Tersoff simulation more similar to the AIREBO case?
In summary, with the Tersoff potential I do not reach the expected results either physically or numerically. I am searching for the cause of this issue—whether it is due to the Tersoff coding itself or if other modifications, such as adding auxiliary commands like fix spring/self, are needed to obtain correct behavior.
I have searched extensively through the LAMMPS manual, related papers, forum threads, and every other source I could think of, but I have not yet found a definitive answer to this ambiguity.
Any help would be highly appreciated. Even a small hint or a keyword to further guide my search would be extremely helpful and valuable.