Hello everyone
As I am new to LAMMPS and molecular dynamics, I am trying to regenerate some data reported in literature with LAMMPS. Specifically I am working with the paper titled ‘An Atomistic Carbide-Derived Carbon Model Generated Using ReaxFF-Based Quenched Molecular Dynamics’ (doi:
C | Free Full-Text | An Atomistic Carbide-Derived Carbon Model Generated Using ReaxFF-Based Quenched Molecular Dynamics).
The first step reported in the paper involves quenching a structure in NVT ensemble. I was able to do that and produced results in close agreement with the paper. The LAMMPS input script is as follows:
units real # for reaxff forcefiled
atom_style charge # for reaxff forcefiled
boundary p p p # 3d periodic boundary
region box_ block 0 74.88 0 74.88 0 74.88 # box dimension 7.488 nm
create_box 1 box_
region whole block INF INF INF INF INF INF units box
create_atoms 1 random 20000 539816 NULL # 20000 atoms are placed at random inside the box
group 1 region whole
neighbor 2 bin
neigh_modify every 1 delay 0 check yes
mass 1 12.0107 # mass of carbon atom
pair_style reax/c NULL # interatomic potential
pair_coeff * * ffield.reax-2013.C C # coefficient for carbon atom
fix 1 all qeq/reax 1 0.0 10.0 1.0e-6 reax/c # for reaxff
thermo 10
minimize 1.0e-4 1.0e-6 100 1000
reset_timestep 0
velocity all create 3500 729014 mom yes rot yes dist gaussian
timestep 0.5 # fs
dump 1 all custom 1000 C.* id xs ys zs fx fy fz
thermo 100
thermo_style custom step time temp density press pe ke etotal
restart 10000 file.*.restart
restart 500 file1.restart file2.restart
fix 2 all nvt temp 3500 3000 10 # tdmap = 10 fs (unit = real = fs)
run 100000 # 50 ps
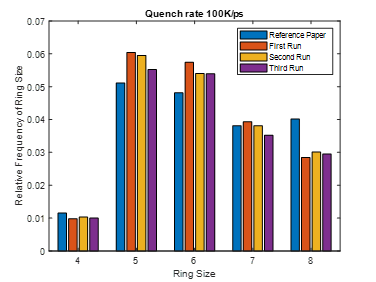
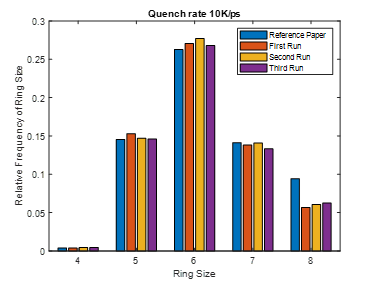
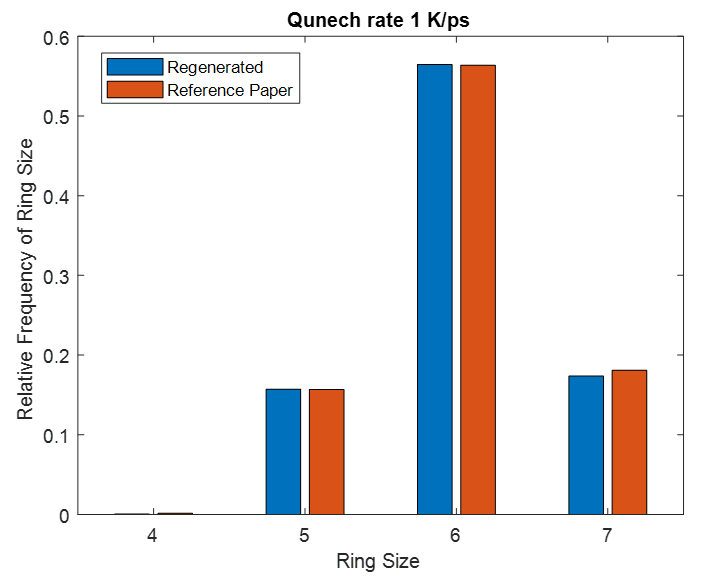
This same input script is run for three different quench steps. The density, cell sizes and ring size distribution for all simulation matches to that of reference paper.
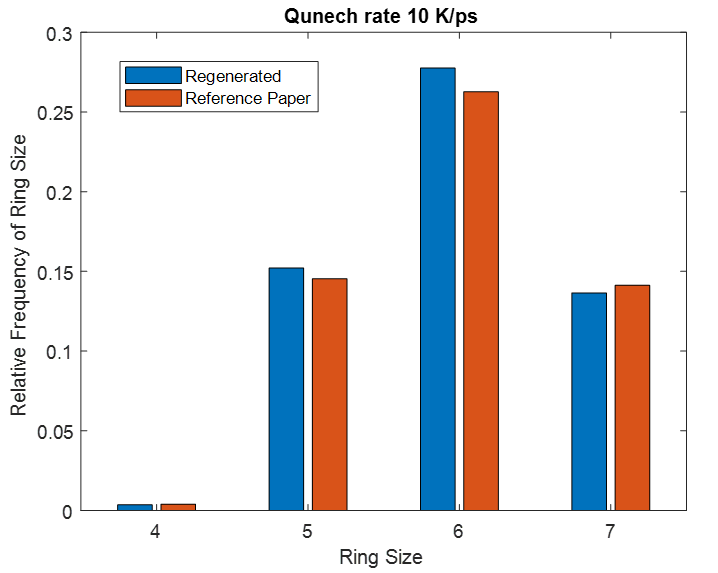
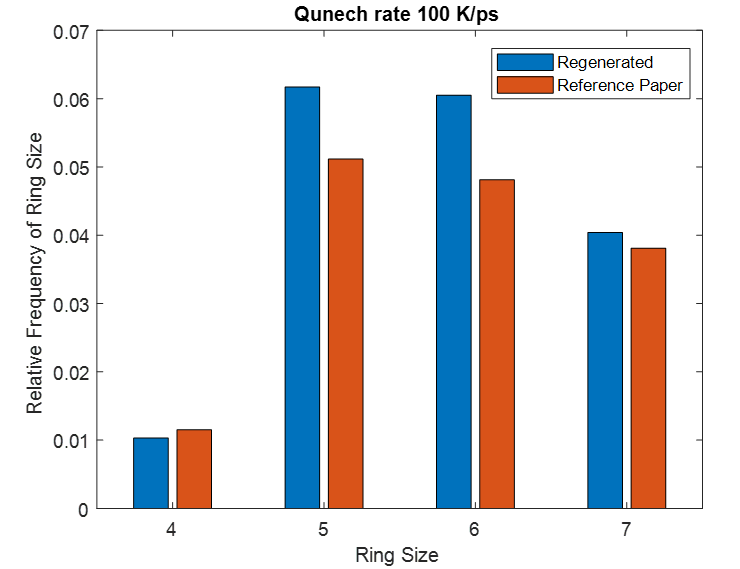
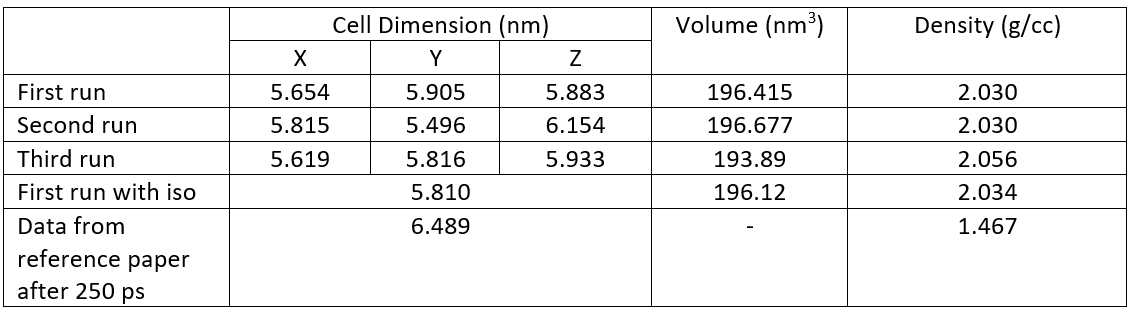
The next step is compressing the previously quenched structure in NPT ensemble. As stated in paper, 'Isothermal temperature control and isobaric pressure control at 3000 K and 20,000 atm were used with damping constants of 10 fs and 100 fs,respectively. I cannot reproduce the data reported in this step.
In the paper it is reported that the final cell size and density for QMD-10c structure will be 6.489 nm and 1.467 g/cc after 250 ps of simulation time. In my simulation, after 52.5 ps the cell size and density appear to be 5.804 nm and 2.0398 g/cc.
For simulating the NPT step, the restart file generated previously was read in and following command line was used:
fix 2 all npt temp 3000 3000 10 iso 20000 20000 100 # tdmap = 10 fs (unit = real = fs) pdamp = 100fs
I cannot figure out how is it possible that the results from NVT ensemble matched but not the results from NPT ensemble? Any help or suggestion is much appreciated.
Regards
Koushik