Dear LAMMPS community,
I mean the configuration integral Q_N in the partition function:
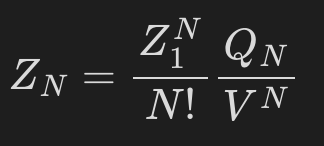
Is it possible to compute it at runtime for given atoms (atom IDs or group) and a specified temperature? What is the way to do it?
Or probably it is possible to compute the whole partition function?
Maybe start here: Extraction of configurational entropy from molecular simulations via an expansion approximation | The Journal of Chemical Physics | AIP Publishing
Please note that the quantity you are thinking about is not trivial to calculate, and you are likely to get stuck without the help of a local, experienced mentor or supervisor.