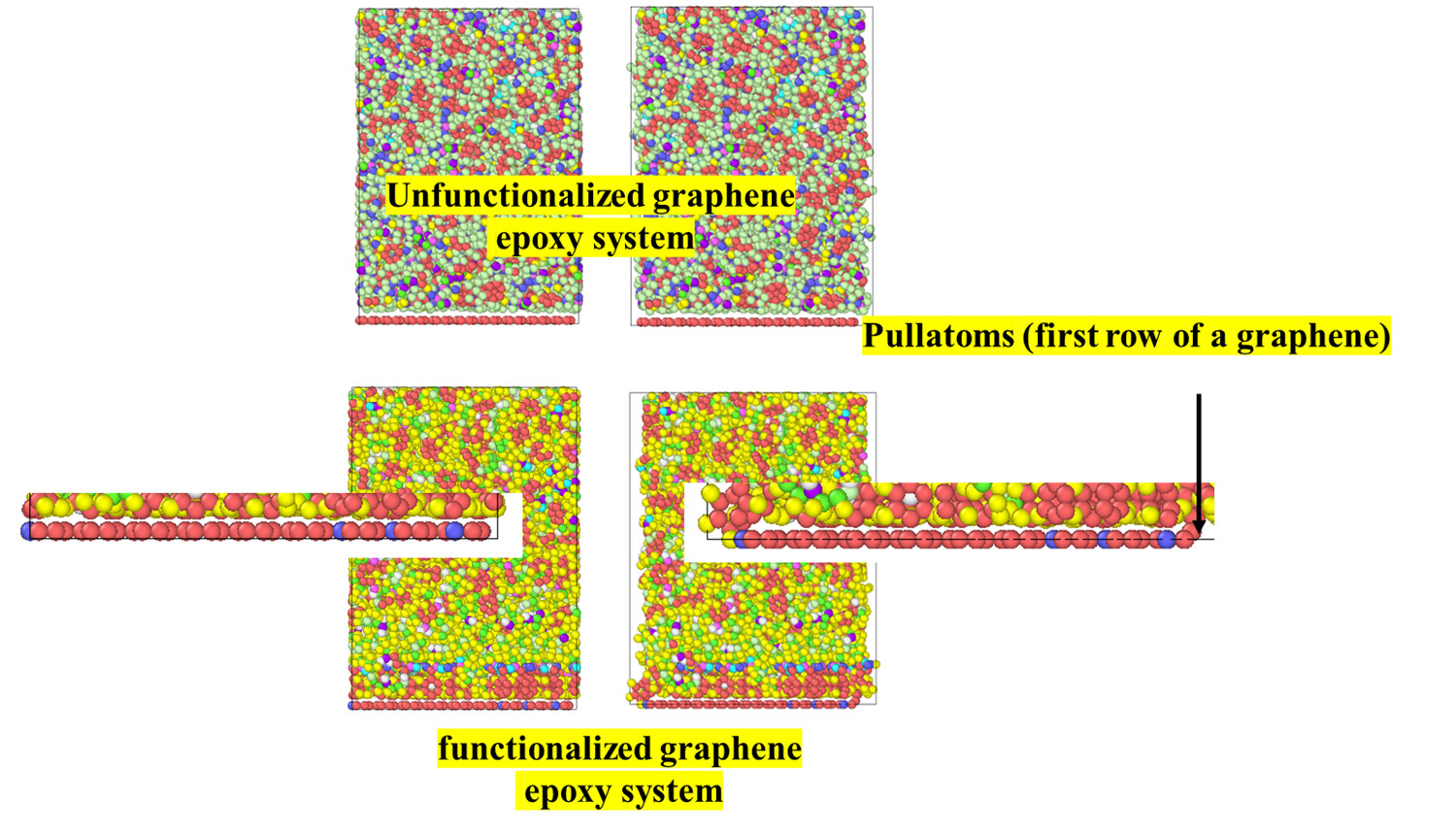
Hello LAMMPS users, I am using windows 30 july 2021. I want to pull a graphene out of epoxy. I am interested in finding change in potential energy with respect to pull-out distance in x direction. Accordingly, boundary conditions are m p m for x,y and z direction.
Before pulling out graphene, I equilibrated structure at 0.01 K.
Top layer of epoxy in Z direction 10 A is kept fixed out of 55A epoxy by not time integrating and named as group polymerfix. Rest of epoxy molecules is assigned group as polymermov.
In case of a graphene, I have assigned a group named as pullatoms for first row of a graphene and kept stationary by not time integrating, and rest graphene atoms are assigned to a group named as rigidgraphene. I do not want movement of graphene atoms in z direction. Therefore, I used command velocity and setforce.
Command used as
velocity rigidgraphene set NULL NULL 0.0
fix fzrigidgraphene rigidgraphene setforce NULL NULL 0.0
fix 2 polymermov nvt temp 1E-8 1E-8 100
fix 3 rigidgraphene rigid/nvt single temp 0.1 0.1 100
I observed in trajectory that atoms of graphene in z direction are in the same direction as z positions are in initial data file. Z positions of atoms only varied by 10.34 to 10.36 A.
But when I used same script for functionalized graphene. I am observing up and down movement in functionalized graphene as shown in Fig. below. How can I make rigidgraphene such as its Z positions doe not change.
Thank you,
Ankit