Dear LAMMPS users,
I want to construct a grain boundary of a simple bulk of BCC iron using LAMMPS. Since I am a beginner, I dont know how to approach this problem. Could you please help me how to do that and what are the parameters should I be considered?
In the simplest case you can split your simulation box in two regions and then use the lattice command with different orientations followed each by a create_atoms command to fill the corresponding region with atoms in that orientation.
For more complex cases, you probably want to look into a pre-processing tool like Atomsk.
I tried to construct for simple bulk of BCC iron in static equilibrium. but not getting right output. still facing errors. Can anyone have a look into this and correct my errors.
#==============INITIALIZATION =======================================================
#Energy:eV Distance:Angstrom Mass:Kg Time:picosec
units metal
#Classical particles
atom_style atomic
#p=Periodic, in x y z
boundary p p p
#lattice constant i.e. Fe-BCC 2.8665
variable a equal 2.8665
#export OMP_NUM_THREADS=12
#BCC structure
lattice bcc $a
#Specify simulation box
region box block 0 5 0 5 0 5
#Initiate box with 1 atom type
create_box 1 box
#Create 1 atom type on lattice in simulation box
create_atoms 1 box
mass 1 55.85
#Interatomic potential - Embedded Atom Method
pair_style eam/fs
#interaction pairs , file name , Element parameters
pair_coeff * * Fe_mm.eam.fs Fe
#Specify build neighbor list, use cut off+0.3 A
neighbor 0.3 bin
#Frequency to rebuild neighborlist
neigh_modify every 20 delay 0 check no
#Compute the energy per atom
#compute eperat all pe/atom
#Output x,y,z of atoms LAMMPS standard format
dump config all atom 10 dump.FeBCC_${a}
#Custom output of atom properties
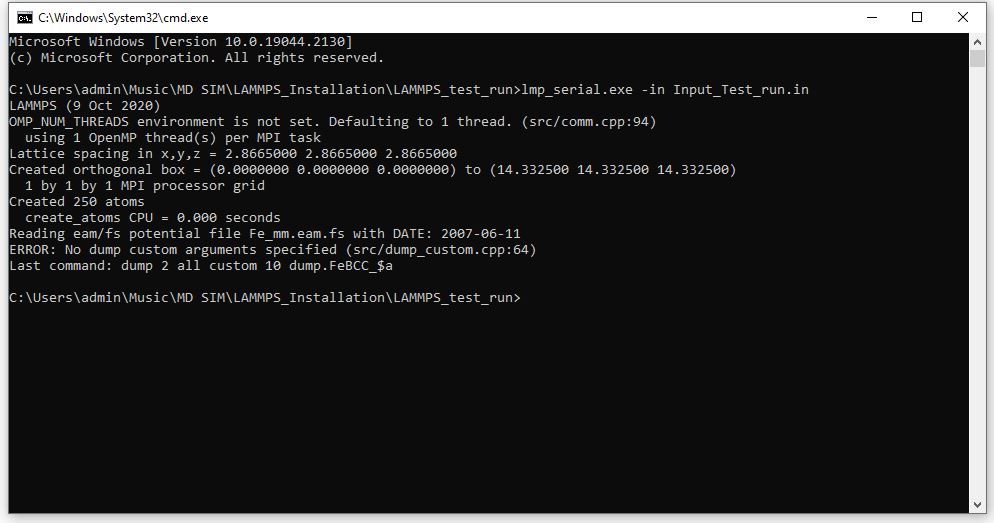
dump config all custom 10 dump.FeBCC_${a} & id type x, y, z #c_eperat
timestep 0.001
#Ensemble
fix 1 all nve
#Frequency of Ensemble data output to screen
thermo 500
#Data that is output to screen
thermo_style custom step pe ke temp vol press
run 10000
What are the errors you’re facing? You need to be more descriptive.
I do notice that your dump command has commas (“,”), which it should not.
Seems to me that the message is clear enough… your dump custom command is lacking the mandatory arguments.