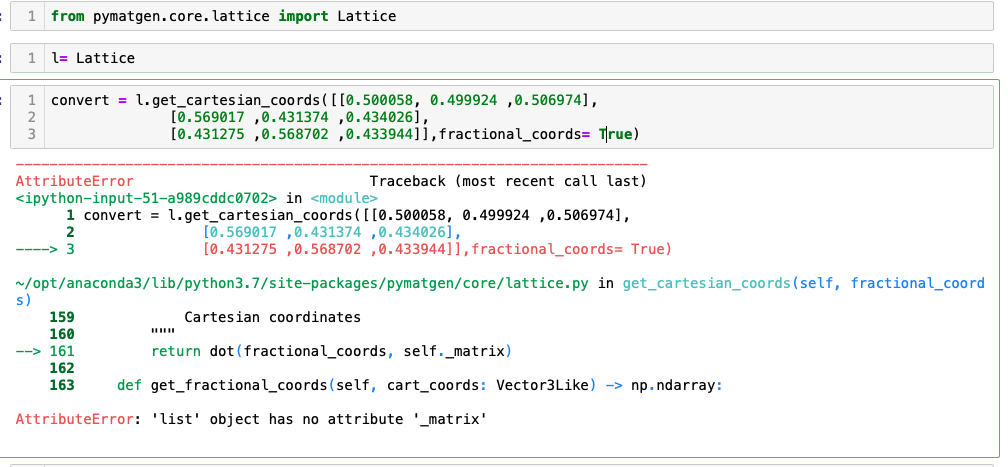
I am trying to convert the fractional cords to cartesian cords so that I can use it in generate_adsorption_Structure in the AdsorbateSiteFinder. Since my VASP relax results returned direct coordination, when I used the output from VASP as input for Molecule to generate and add the adsorbate in to my slab, the adsorbate overlapped together into one atom. I read the docs and I think the reason is because the Molecule read the input in cartesian cords, and my input is in fractional cords. I found the get_cartesian_coords from Lattice module and tried to convert fractional to cartesian but it kept showing error and asked for fractional_coords = as below
The point is, you have not initialized your Lattice object yet. You can look at the source code in pymatgen/core/lattice.py at init method. You have to provide three vectors that define your Lattice. For example,
Thank you for the explanation. I will give it a try. I also found that if I called .cartesian_coord from Structure, I could get the coordinates too. Just not so sure if they are correct
No, it does not. The init method of Structure class in pymatgen/core/structure.py has a variable coords_are_cartesian: bool = False/True which just clarifies that the “coords” you use to initialize Structure are given in Cartesian or fractional coordinates.
Structure contains site objects. For example, you have structure object (defined through Structure class), then try
sites = structure.sites
for site in sites:
print(site.coords)
print(site.frac_coords)
where, site.coords and site.frac_coords give you Cartesian and fractional coordinates, respectively.
Sorry for a misunderstanding. I don’t remember the Structure has cartesian_coord attribute as you mentioned but you can try and compare with the coordinates from site objects.
I just tried both, and I got the same coords. So happy to solve this one.
One thing for the for-loop is that if we print out both frac and cartesian, the coords will be sorted.
Thank you again!
Ngan