Hello,
I have a question about converting units from "real" to "metal".
I've read similar kind of posts but I'm still wondering.
I converted units from "real" to "metal" in the sample input file in the
document of "compute heat/flux".
I use the same random number seed, and at the time step 0, the
calculated total energy is the same.
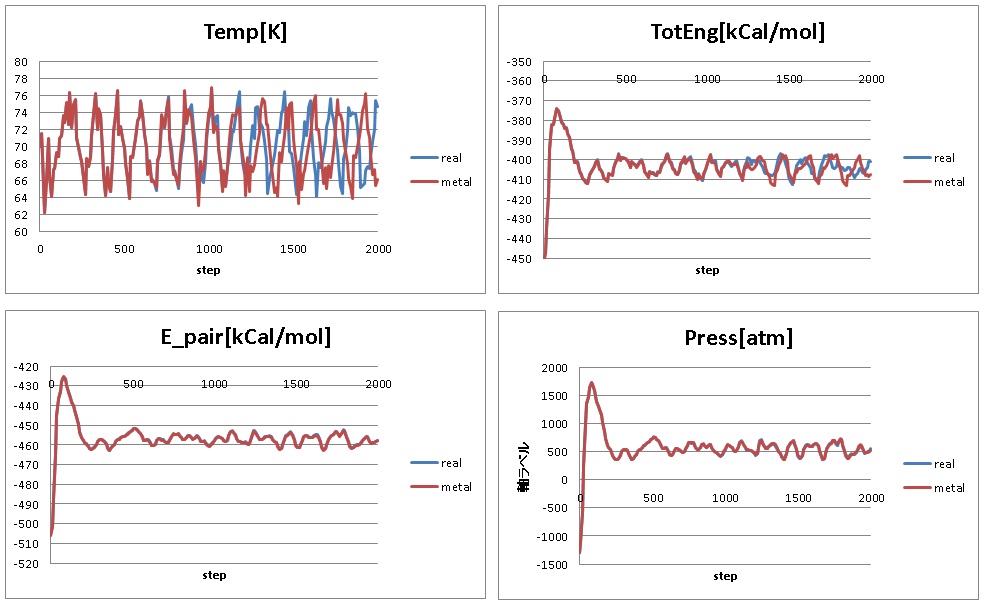
However, at timestep 2000, they have different temperature,
74.682256[K] from the "real" units input file and
66.078762[K] from the "metal" units input file.
and the calculated thermal conductivity have different result,
0.29[W/mK] from the "real" units input file and
0.19[W/mK] from the "metal" units input file.
Where are the differences from?
Anyone has any ideas?
My conversion process is below.
1. I converted values which have units of time[fs] to [ps] in the
definition of "dt" and "fs2s" and the Tdamp value in fix nvt.
2. I converted values which have units of energy[kCal/mol] to [eV] in
the definision of "kCal2J" and the 1st potential parameter in "pair_coeff".
3. I didn't change values which have units of temperature[K],
distance[Angstroms] and mass[g/mol] because "metal" and "real" has the
same units of them.
I will attach the both input file of "real" units and that of "metal"
units I used. (The run time is shortened from the original sample file)
#### converted input in "metal" units from here ####
# Sample LAMMPS input script for thermal conductivity of solid Ar
units metal
variable T equal 70
variable V equal vol
variable dt equal 0.004
variable p equal 200 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
# convert from LAMMPS real units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable eV2J equal 1.6021764E-19
variable A2m equal 1.0e-10
variable ps2s equal 1.0e-12
variable convert equal \{eV2J\}\*{eV2J}/\{ps2s\}/{A2m}
# setup problem
dimension 3
boundary p p p
lattice fcc 5.376 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region box block 0 4 0 4 0 4
create_box 1 box
create_atoms 1 box
mass 1 39.948
pair_style lj/cut 13.0
pair_coeff * * 0.0103299 3.405
timestep ${dt}
thermo $d
# equilibration and thermalization
velocity all create $T 102486 mom yes rot yes dist gaussian
fix NVT all nvt temp $T $T 0.01 drag 0.2
run 2000
# thermal conductivity calculation, switch to NVE if desired
#unfix NVT
#fix NVE all nve
reset_timestep 0
compute myKE all ke/atom
compute myPE all pe/atom
compute myStress all stress/atom virial
compute flux all heat/flux myKE myPE myStress
variable Jx equal c_flux[1]/vol
variable Jy equal c_flux[2]/vol
variable Jz equal c_flux[3]/vol
fix JJ all ave/correlate $s $p d &
c\_flux\[1\] c\_flux\[2\] c\_flux\[3\] type auto file J0Jt\.dat ave
running
variable scale equal {convert}/${kB}/$T/$T/$V*s\*{dt}
variable k11 equal trap(f_JJ[3])*\{scale\}
variable k22 equal trap\(f\_JJ\[4\]\)\*{scale}
variable k33 equal trap(f_JJ[5])*${scale}
thermo_style custom step temp v_Jx v_Jy v_Jz v_k11 v_k22 v_k33
run 10000
variable k equal (v_k11+v_k22+v_k33)/3.0
variable ndens equal count(all)/vol
print "average conductivity: $k[W/mK] @ T K, {ndens} /A^3"
#### converted input in "metal" units till here ####
#### input in "real" units from here ####
# Sample LAMMPS input script for thermal conductivity of solid Ar
units real
variable T equal 70
variable V equal vol
variable dt equal 4.0
variable p equal 200 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
# convert from LAMMPS real units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable kCal2J equal 4186.0/6.02214e23
variable A2m equal 1.0e-10
variable fs2s equal 1.0e-15
variable convert equal \{kCal2J\}\*{kCal2J}/\{fs2s\}/{A2m}
# setup problem
dimension 3
boundary p p p
lattice fcc 5.376 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region box block 0 4 0 4 0 4
create_box 1 box
create_atoms 1 box
mass 1 39.948
pair_style lj/cut 13.0
pair_coeff * * 0.2381 3.405
timestep ${dt}
thermo $d
# equilibration and thermalization
velocity all create $T 102486 mom yes rot yes dist gaussian
fix NVT all nvt temp $T $T 10 drag 0.2
run 2000
# thermal conductivity calculation, switch to NVE if desired
#unfix NVT
#fix NVE all nve
reset_timestep 0
compute myKE all ke/atom
compute myPE all pe/atom
compute myStress all stress/atom virial
compute flux all heat/flux myKE myPE myStress
variable Jx equal c_flux[1]/vol
variable Jy equal c_flux[2]/vol
variable Jz equal c_flux[3]/vol
fix JJ all ave/correlate $s $p d &
c\_flux\[1\] c\_flux\[2\] c\_flux\[3\] type auto file J0Jt\.dat ave
running
variable scale equal {convert}/${kB}/$T/$T/$V*s\*{dt}
variable k11 equal trap(f_JJ[3])*\{scale\}
variable k22 equal trap\(f\_JJ\[4\]\)\*{scale}
variable k33 equal trap(f_JJ[5])*${scale}
thermo_style custom step temp v_Jx v_Jy v_Jz v_k11 v_k22 v_k33
run 10000
variable k equal (v_k11+v_k22+v_k33)/3.0
variable ndens equal count(all)/vol
print "average conductivity: $k[W/mK] @ T K, {ndens} /A^3"
#### input in "real" units till here ####
Thank you in advance.
TADA Masahiro