I’ve simulated (for simplicity’s sake) a set of monosized spherical particles with diameter 1 (unitless) within a triclinic box from which I dump a file containing pairwise contacts and their interparticle distance:
compute indicies all property/local patom1 patom2 cutoff radius
compute distance all pair/local dist cutoff radius
dump 2 all local ${stepsize} dist_IJ.txt c_indicies[1] c_indicies[2] c_distance
I wish to study the location of the contact of each particle, i.e., given some particle, how are the contacting particles placed around it?
Some particles are in contact with particles at the opposite side of the triclinic box (as intended), but I am interested in what the coordinate of those reflected particles would be, were they not reflected (i.e. placed outside the boundary of the box). I followed the steps at 8.2.3. Triclinic (non-orthogonal) simulation boxes — LAMMPS documentation in order to transform the particles, see my reasoning below:
After relocating the particle its distance does not coincide with the interparticle distance provided by the dumpfile, which for all particles are approx. 1 since they are just in contact. Instead, the particles seem to overlap, and to a varying degree even though the interparticle distance always should be 1.
Ultimately, the end goal is to identify whether all contacts coordinates to a particle can be contained within the same 3D-hemisphere of the centerparticle. If this condition is met then the particle can be classified as a rattler particle.
So basically the issue is, my method of relocating the reflected particle to its “real” position is wrong, but I cannot identify where. Am I doing it wrong? Is there a simpler way?
Please have a look at 4.4. Parallel algorithms — LAMMPS documentation or the corresponding section of the latest LAMMPS paper. In LAMMPS you don’t have minimum image conventions, but periodic images are realized as ghost particles.
So what does this mean in practice? As I understood it, atoms which are beyond the box boundaries within some cutoff distance are stored as ghost atoms, which are then communicated to another processor, which then calculates its relocated coordinate.
I don’t know if its even possible to dump/store/output the ghost atom info in any way, but even doing so seems complicated and I assume it would slow down the script.
What if I set the cutoff distance of the boundaries to the maximum particle diameter? In the monosized case its simple, although I intend to simulate very polydisperse systems. Then the particle would only relocate once it has fully passed the boundary, and calculations might be more precise. Would this affect the runtime negatively? Could this work?
Actually the interparticle overlap doesn’t really matter anyways, if the relative angle between the centerpoints is the same as in the case where they are placed correctly. But I have no way of verifying that this is the case…
Sorry for bombarding you with questions, got a bit desperate for solutions.
Not box boundaries, sub-domain boundaries. Periodic boundaries are just a special case of the domain decomposition.
This kind of processing will be slow however you slice it when you do it outside a proper programming language and because all operations in the LAMMPS input are global operations and thus non-parallel and often require communication of distributed data, once this becomes an issue of per-atom information.
What you describe seems rather complex and sounds like it would need to be implemented as a compute style.
Not sure if this is helpful, but you can force the dump file to remap particles back into the box, see the “pbc” option in dump_modify: dump_modify command — LAMMPS documentation
“The pbc keyword applies to all the dump styles. As explained on the dump doc page, atom coordinates in a dump file may be slightly outside the simulation box. This is because periodic boundary conditions are enforced only on timesteps when neighbor lists are rebuilt, which will not typically coincide with the timesteps dump snapshots are written. If the setting of this keyword is set to yes , then all atoms will be remapped to the periodic box before the snapshot is written, then restored to their original position. If it is set to no they will not be. The no setting is the default because it requires no extra computation.”
Thank you for the reply. I added the pbc keyword to a dump modify which dumps coordinates. It did not result in the the behavior I had hoped. The remapping of atoms which are partially outside the box did not relocate them to the “unreflected” coordinate.
I tried to collect the unwrapped coordinates through “compute uw all property/atom xu yu zu” and dumping them together with the wrapped coordinates x, y, z (I guess they’re called?).
In the cases where a mismatch was found, i.e. pairwise distance from dumpfile was not same as calculated euclidean distance by positions, the unwrapped coordinate proved to be correct, but not always. In some cases both the unwrapped and wrapped coordinates were still wrong.
Can it be that in the cases when neither wrapped nor unwrapped coordinates are correct, it is because the particle has not gone beyond the cutoff distance of the periodic boundaries? So it’s placed a bit outside of the periodic boundary such that it is in contact with a particle at the opposing side, but not far enough that the unwrapped coordinate is the reflected version of the wrapped one?
Its a bit disheartening because at first I thought I had found a solution. But it atleast reduces the number of reflection cases to handle.
If any of you still feel motivated to help me it’d be appreciated!
It would help if you could make a small working example showing us exactly what you want and what LAMMPS is printing out instead. I’m not familiar with your field or your work and so it’s very difficult to even visualise your exact situation.
It works now. I misinterpreted the input for the matrix I previously denoted as “H”, described at 8.2.3. Triclinic (non-orthogonal) simulation boxes — LAMMPS documentation . I used ‘xlo_bounds’, ‘xhi_bounds’, … etc supplied from dumpfile at each timestep as ‘xlo’, ‘xhi’, … i.e. the bounds used to transform the coordinates to unwrapped were using the bounds of the orthogonal box which encloses the triclinic box, thus resulting in the incorrect position.
The above mentioned reasoning is used and works for all my cases.
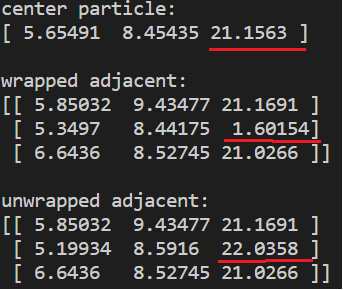
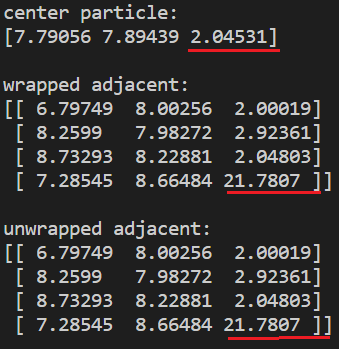
I might have been a bit unclear in describing the issue at hand, as srtee mentioned. Here are two examples which might make it a bit easier to visualize. The images show R3 coordinates of the center particle and its adjacent (contact) particles for both x,y,z (wrapped) and xu, yu, zu (unwrapped). All particles have equal size (diameter 1) and adjacent particles are placed at an euclidean distance 1 from the center particle.
This is an example where the unwrapped coords yields the result I want. The wrapped coordinate is obviously not placed at euclidean distance 1, but the unwrapped one is.
This example shows a case where neither the unwrapped nor wrapped coordinates are at euclidean distance 1 from the center particle.
Since cases of the last example occur sometimes, I resort to just finding the n* as described above.
Thank you all for the help, it’s a luxury having a forum like this!