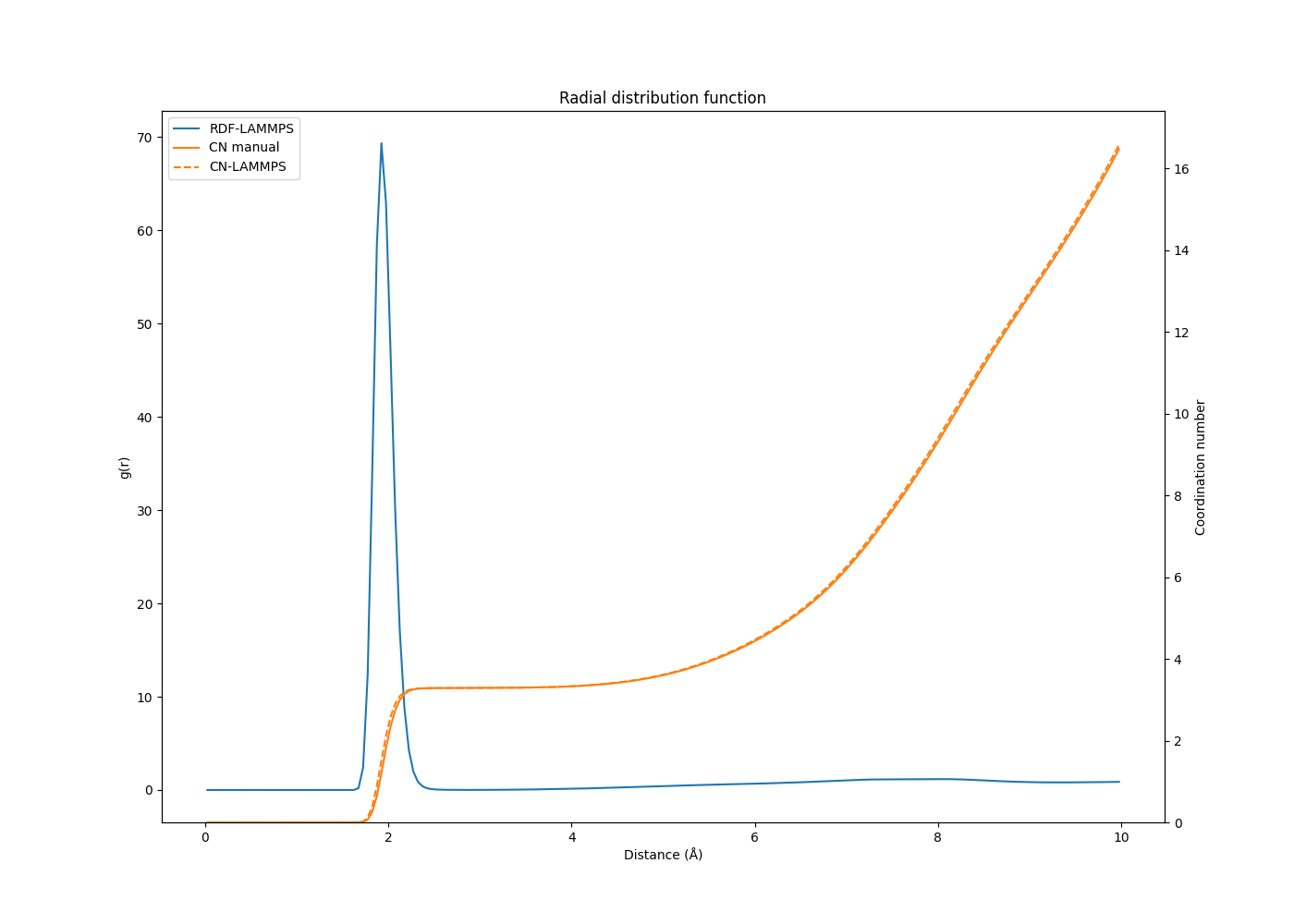
It seems to me that the coordination number calculation when I use compute rdf in this particular simulation is wrong.
When I calculate the CN myself from the rdf output by LAMMPS, I get different values than the CN from LAMMPS, and the values seem to be off by some factor.
Code example of how to calculate CN:
from scipy import integrate
import math
# store LAMMPS RDF from rdf.dat as variable RDF, store centers of bins as variable centerbin, store box volume as variable volume
CN = integrate.cumulative_trapezoid(RDF*4*math.pi*centerbin**2,centerbin, initial=0)*182/volume
# 182 is here the number of jtypeN, so 182/volume should be the global average volume density of jtypeN.
I have attached the start configuration restart_eq.data and parameters system.settings, along with the input file rdf.lmp. The output rdf from the simulation is rdf.dat. The LAMMPS version is LAMMPS (29 Aug 2024). The simulation is run on a cluster with Intel Xeon Gold 6348 CPUs and Rocky Linux release 9.4 (Blue Onyx).
Would be great if someone could double check that I am not doing a mistake in my CN calculation (and try to reproduce the data I get here). Here are the LAMMPS RDF, LAMMPS CN and the calculated CN (CN manual) plotted together:
I can note finally that the calculated CN becomes similar to the LAMMPS CN if I multiply by the factor 14/volume instead of 182/volume. 14 is here the number of itypeN (central atom type) so that would be wrong, I think.
The coordination number in LAMMPS is computed directly from summing over the histogram collected for computing the g(r), and thus by construction it is very accurate, much more accurate than it can ever be from (numerical) integration. Please note that the LAMMPS g(r) output is corrected for finite size effects (if not its limit is not 1.0 but (n-1)/n with n being the number of pairs of particles.).
This has been tested many times and also the exact same approach has been implemented into VMD where it also has been tested many times over many years.
Thus I am very confident, that - if there is a problem - it is likely with your code and not LAMMPS.
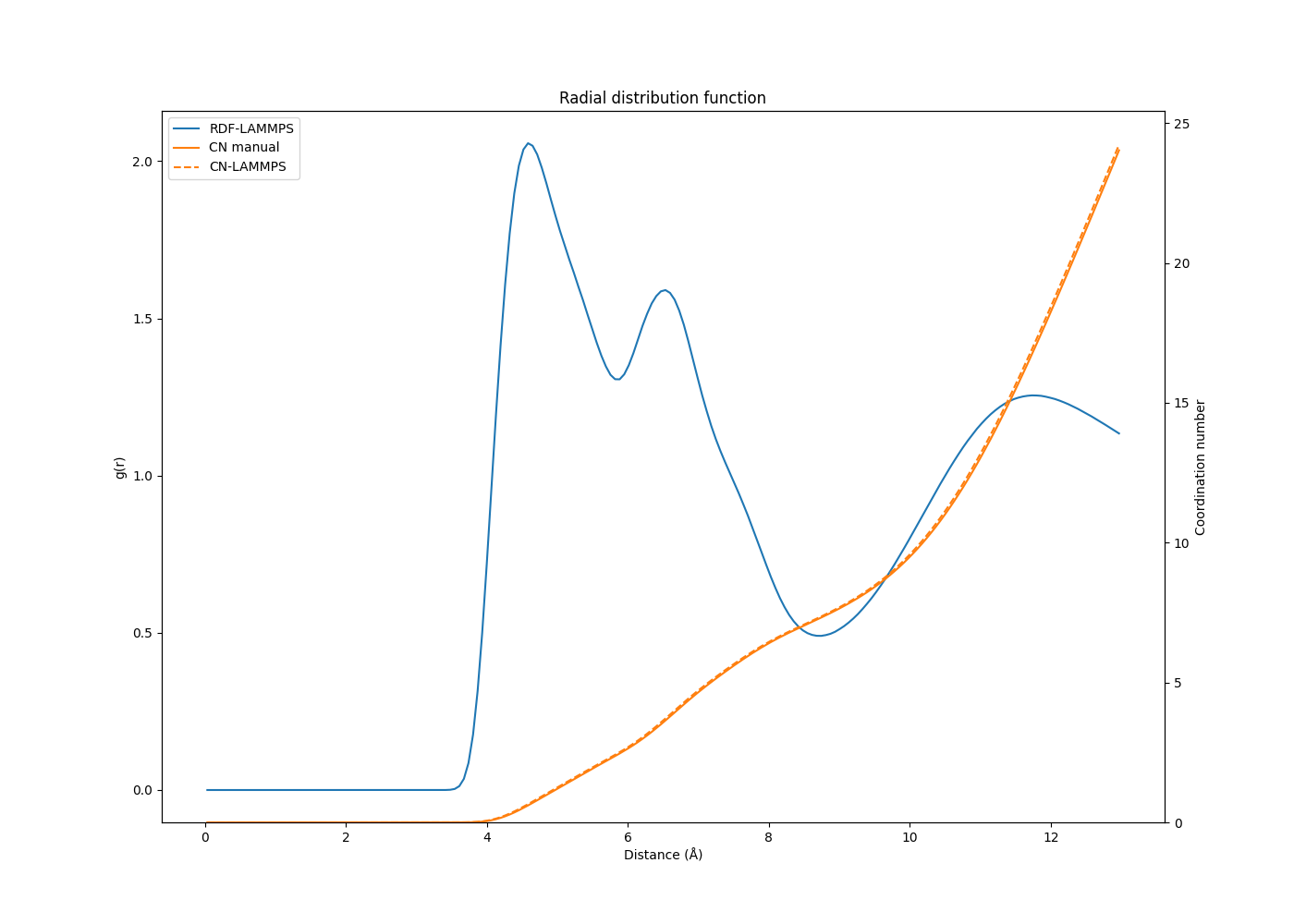
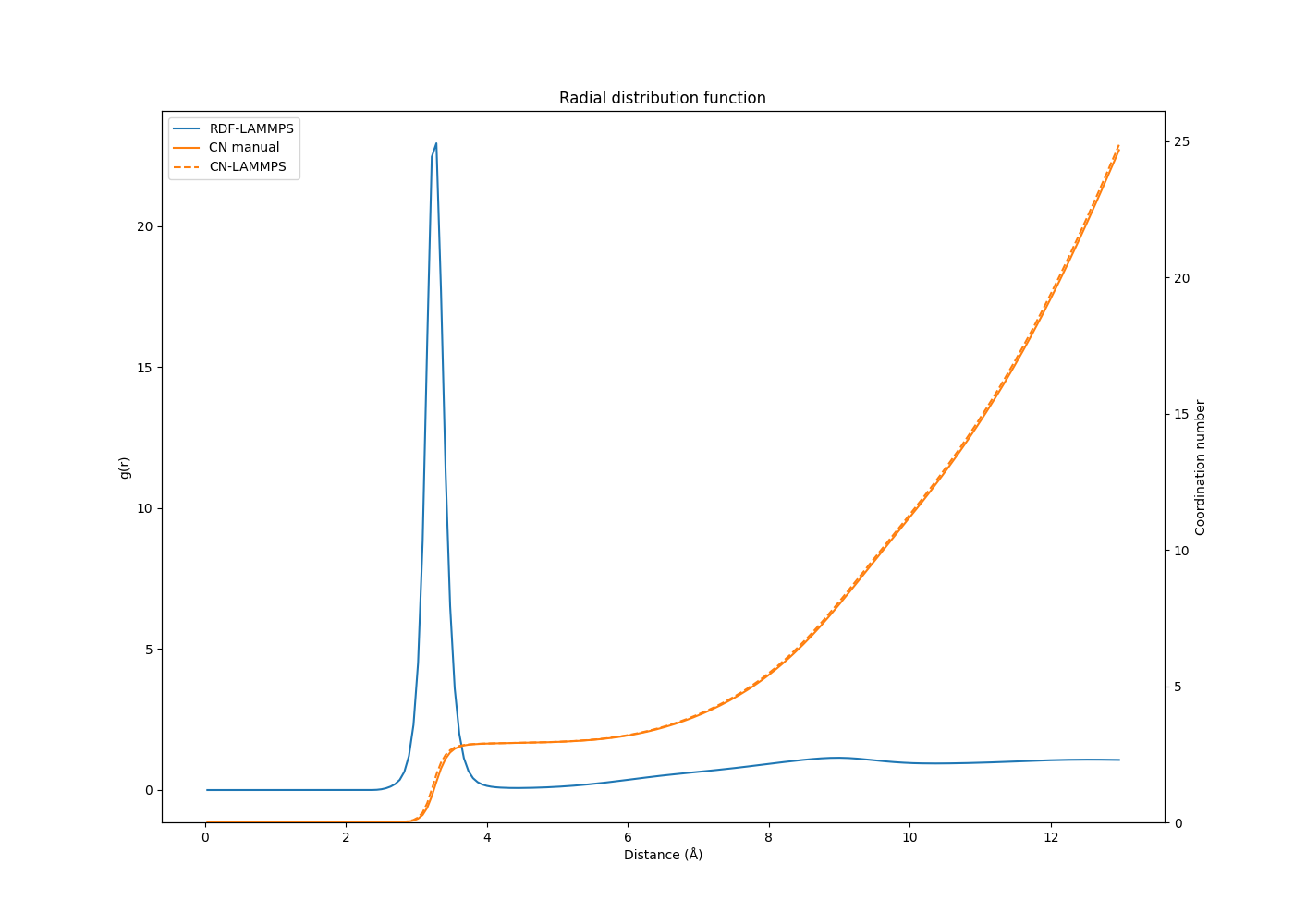
What bugs me is that when I calculate the CN by numerical integration of the RDF data from other simulations, I obtain very similar results to the CN from LAMMPS. Of course the numerical integration is not as exact as the LAMMPS calculation, but when the bin size is small the results almost overlap. Some results underneath:
The first RDF/CN plot is from a system which is similar (same number of atoms and molecules) to the RDF plot from my initial post, except the potential used is different. This is what made me react to those results in the first place.
It is understandable if you don’t want to spend time on this potential non-issue (there is a relatively high probability I made a mistake here, after all), but if someone would please rerun the simulation and check the results I would get some peace of mind, if nothing else.