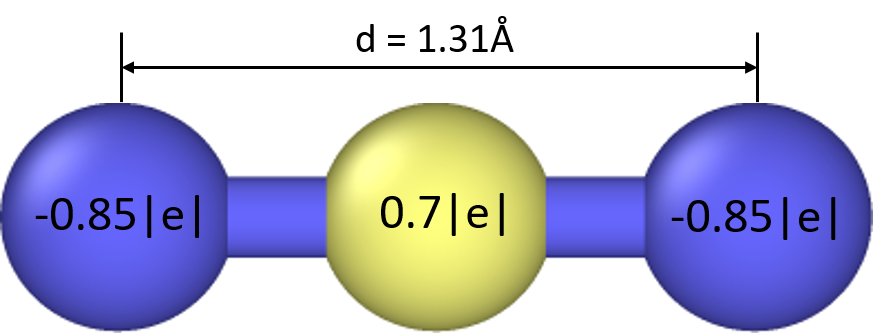
I’m a new GULP user. I’m trying to calculate phonon DOS plots for NaO2 supercell - in which bonded oxygen ions are modelled as rigid bodies. Additionally a particle (dummy) having charge 0.7 and mass 0 is also introduced at center of mass of superoxide molecule. I’ve attached an image of oxygen model - blue spheres refer to oxygen atoms with -0.85 charge and yellow sphere is a massless COM particle with 0.7 charge. So net charge of one superoxide molecule is -1.
I used “connect” command to specify the bonds between these particles along with image flags of second atom (input gulp file attached). But I still seem to be getting coulomb subtraction error.
Any ideas what is happening to cause this?
PS: I’m using buckingham potential with coeff as specified in input file.
You need to add the “noauto” keyword to your input. By default if you specify “molecule” GULP will try to compute the connectivity automatically based on the covalent radii. So if you want to specify it manually with “connect” you need to turn this off with “noauto”.
Thanks a lot for your quick response. It was very helpful.
I’ve a quick follow up question - I’m currently using two connect commands (for one superoxide molecule) to specify the bond between left oxygen with COM and COM with right oxygen atom (as shown in picture). Is this a right modelling in GULP - which still treats it as one single linear molecule and neglects all the columbic & buckingham interactions between left and right oxygens i.e. only intermolecular interactions are included?
Also, since these particles are treated as point masses (with charge) in LAMMPS, should I explicitly specify covalent radii of these particles to be 0 ?
PS: I believe using “molecule” and “inter” removes intramolecular interactions between left and right oxygens with COM. I’ve attached my updated input file for your reference.
That all sounds correct. You can always test by running a molecular calculation to see that the intramolecular energy is zero. No need to set the covalent radii to 0 as these are only used when determining the bonding and so as you have noauto then this makes the value redundant.
I tried commenting out “inter” in the above input GULP setup. I observed different interatomic potential energy this time than previous (i.e. with inter).
If GULP treated superoxide molecules as single linear rigid body previously, can you please explain why do I observe this change between with/without inter?
More details; From help docs, I understand that “inter” treats all subsequent potentials, here buckingham, as intermolecular when molecule option is active. So, in case of rigid bodies, this shouldn’t be affecting right? Unless {left oxygen - COM} and {COM - right oxygen} are treated as two separate rigid bodies, rather than one single rigid superoxide molecule…
Hi Erick,
Just because a body is rigid, doesn’t mean to say that it can’t have an intramolecular energy. It’s just that it will be a constant due to the fixed geometry. Commenting “inter” will just mean that you shift the energy within the molecule but won’t change the intramolecular geometry.
Cheers
Julian
I have another follow up question - I tried to verify the GULP input setup file by comparing lattice energy calculated from GULP with potential energy (PE) calculated from LAMMPS (i.e. with the PE for the same snapshot at t=0 step from LAMMPS) Interestingly though total PE seem to be matching, almost exactly, across LAMMPS and GULP when both potentials are turned on, individual components seem to be not matching.
By turning off coulombic interactions in LAMMPS (i.e. by using just “pair_style buck”) I ran rigid body simulation and calculated potential energy at t=0 step. I compared this value with the “interatomic potential” component in GULP (available under “Components of energy” section). Surprisingly this was different! To be precise, interatomic potential from GULP in around 1211 eV whereas PE from LAMMPS is around 242 eV.
Similarly to test coulombic values, I turned off buckingham potential in LAMMPS (by using just “pair_style coul/long”) I ran rigid body simulation and calculated potential energy at t=0 step. I compared this value with the “Monopole - monopole (total)” in GULP (available under “Components of energy” section). Again, this was different too.
So, can you please share your thoughts on these differences? Is it reasonable to expect these component values to be around same magnitude.
I’m sorry for asking many questions. Would be really great if you can help me out.
PS: I use the same cutoff for buckingham potential in LAMMPS and GULP. I also use same real/reciprocal space cutoff for ewald sum in LAMMPS and GULP. I implement real/reciprocal cutoff in GULP using “ewaldrealradius”.
If the total potential agrees then that’s what you want. When you have molecular Coulomb subtraction going on (especially with 1-4 etc) then how some terms are partitioned between the two-body/pair energy & Coulomb can get muddied. In GULP, the electrostatic terms are those that come from the Ewald sum (or similar), while the Coulomb subtractions are implemented as a pair potential & so end up elsewhere.
Hope that explains the differences.
Thanks for you reply and sorry for my late response. I was trying out different methods to calculate phonon density of states, assuming the current setup is correct.
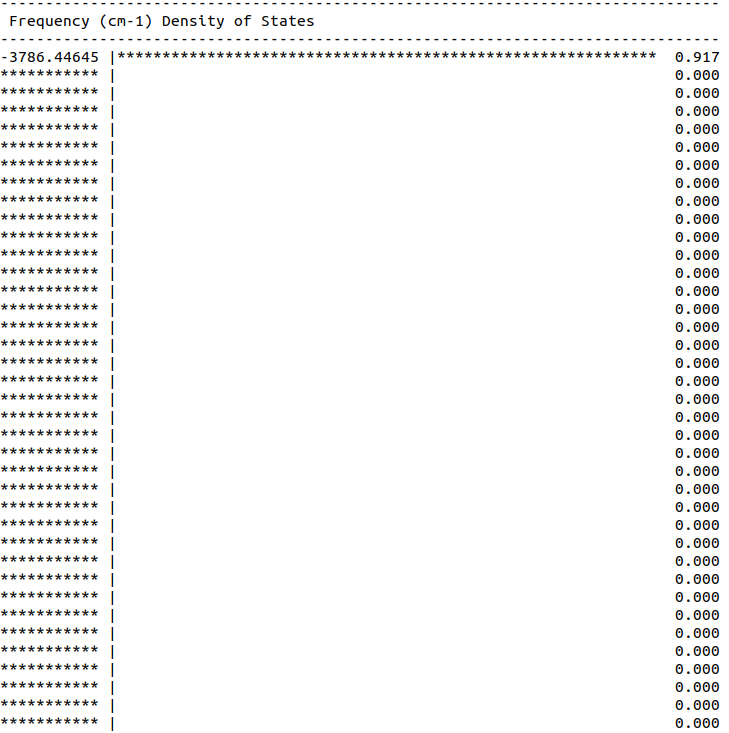
But unfortunately I wasn’t able to get any reasonable values, i.e. I always ended up with some “*” like shown in the attached plot.
Details of this plot: I used an unit cell of NaO2 with 4 Na and 4 superoxide molecules (i.e. with 8 Oxy atoms and 4 dummy He atoms at COM of oxygens). I followed the same setup as previous (attached here too). I also tried relaxing the structure in GULP before doing DOS calculations (using keywords in line 2), but no luck there. So would be really great if you can share your thoughts on this setup/calculations?
The reason for this is that if you look at the output from the run you’ll see you have some crazy frequencies - you have imaginary modes of order -3600 cm^-1 and several frequencies that are too large to print & so the whole scale is messed up. Bottom line is that the force field is no good & you have nonsense frequencies, so you’re not going to get a good PDOS until you fix the force field.
In this authors have compared dispersion plots generated using this force field with experimental values (as shown in figure 1 of the above attached paper) - which seem to be matching well. So I followed the same parameters and potential form to replicate the curves using GULP. But unfortunately I have been getting high freq values.
Can you please let me know if I’m missing something here?
I’m sure these authors did obtain good results for this system. Of course you’ll only get them if your input reproduces their force field and then you’ll get the same set of answers. At the moment there seems to be something different in your input from the paper in question.
Thanks again for replying and sharing your views. I compared my setup with author’s.
I tried replacing mass of dummy He atoms from 1e-7 to exact zero, keeping the rest same (attached is the modified input file). Surprisingly this gave reasonable DOS plots and dispersion curves. I was expecting it to crash like in LAMMPS which gave an error of “invalid mass value”, since the mass was exact zero. But GULP seem to be interpreting it in a different way.
I tried looking up some similar examples/in manual, but no luck. Can you please share your thoughts on what could be happening here? Sorry for asking many questions, but this seems to be very different!
I can certainly comment on the mass when it’s zero. Because the atom is part of a rigid molecule, what matters is the total mass of the molecule. If this was zero then things would go horribly wrong, but the mass of an atom within it can be zero.
NB Although the frequencies are better than for your previous inputs, the elastic constant tensor is very ugly and there are lots of imaginary modes away from gamma, so there’s still some work to be done to get a sensible model.
Thanks for sharing your thoughts! This was really helpful in getting intuition about zero mass.
I think this was due to img flags of “He” atoms at the box boundary. In LAMMPS, “He” atoms at the boundary take img flags of 1 (based on dump file). But in GULP I think these “He” atoms should be assigned 0 for the bonding to be made correctly. Modifying the img flags of corresponding bonded “He” atoms seem to fix this imaginary values. I’ve attached the modified file for an unit cell. PFA
With this change, the energy minimization also converges in one step. This shows input structure itself is already relaxed - which is what I was hoping for.
But I noticed in GULP output file that elastic constants along 11, 22, 33 components are not equal, contrary to the structural symmetry. I seem to get them equal in LAMMPS though. So, am I missing out something on specifying symmetry in the input structure/file? Can you please help me out.
Hi Erik,
I’ve had a quick look into this and I think there must be a code issue connected with the rigid molecules and your particular setup. It appears that if you add the keyword “prop” to explicitly generate the elastic constant tensor then you get a sensible set of values (though there seems to be something not quite right with the dielectric constants). Normally if “phon” is specified then this triggers “prop” by default, but for rigid molecules there must be a small code logic issue such that this isn’t happening early enough to get the settings right. I’ll look into how to fix this, but in the meantime just add “prop” to the keyword line.
Regards, Julian
I’ve now found the problem - the value of “lstr” needs to be set to true in setup.F90 based on lphon as well as lprop & then this fixes the problem for the “prop” or not issue. This fix will appear in the next patch release.
PS I notice you have tab characters in your input - I’d avoid these since they can cause problems with input parsing where a space is looked for.