Dear developers and users,
I am comparing the same MOF model with different bond energy terms.
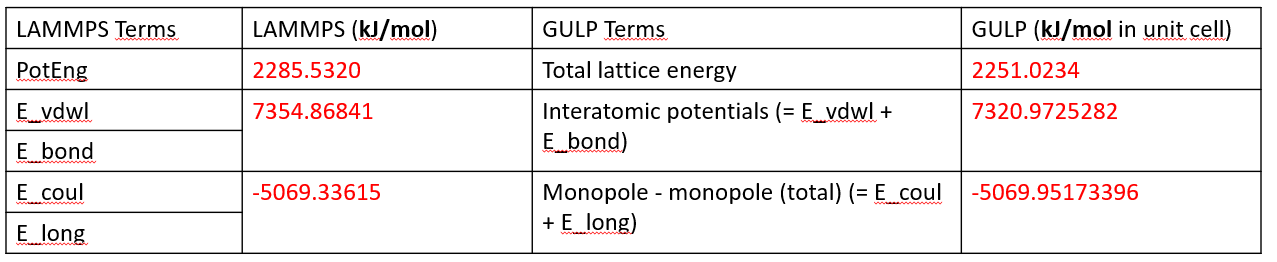
in the first case, there are 4 types of LJ parameters and 6 types of bond terms. the energy calculation was implemented in LAMMPS and also validated using GULP. Both the interatomic terms and coulombic energy are well-matched in the two software.
in the second case, I only removed one bond term (i.e., Cu-Cu bond term that in reality does not exist), and kept others the same as in the first case. The interatomic terms were well-matched with the one from GULP, but the coulombic energy differs by 873.68 KJ/mol between the two programs. and the one from gulp is similar to the first case, which I think makes sense since both the framework and charges were kept the same for the 2 cases.
here is the energy comparison for 2 cases by LAMMPS, the coul energy differs by ca. 200 kcal/mol when the framework and charges are almost same:

so I think the problem probably occurs in my lammps input file. Only removing one bond term caused such a large difference in coulombic energy makes me confused.
here is the comparison between lammps and gulp for 2 cases:
4 LJ+6 bonds:
4 LJ+5 bonds:
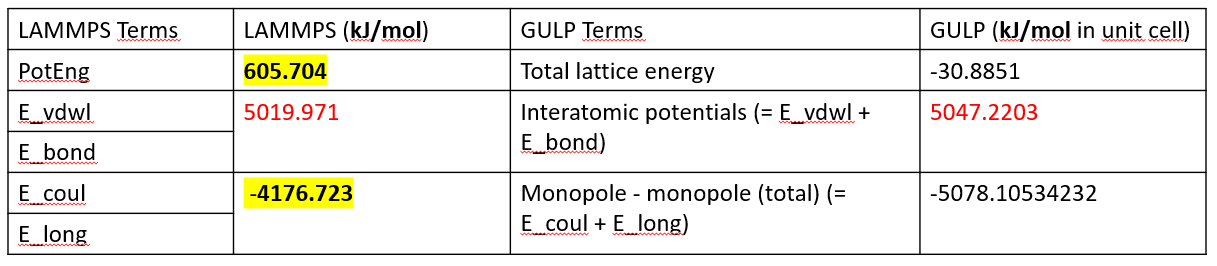
can anyone spot what’s wrong with my calculation? thank you in advance.
4lj+6bonds.Cu-Cu (45.8 KB)
4lj+5bonds.no_Cu-Cu (60.0 KB)
in.4lj+5bonds (569 Bytes)
in.4lj+6bonds (568 Bytes)
Best regards,
wang