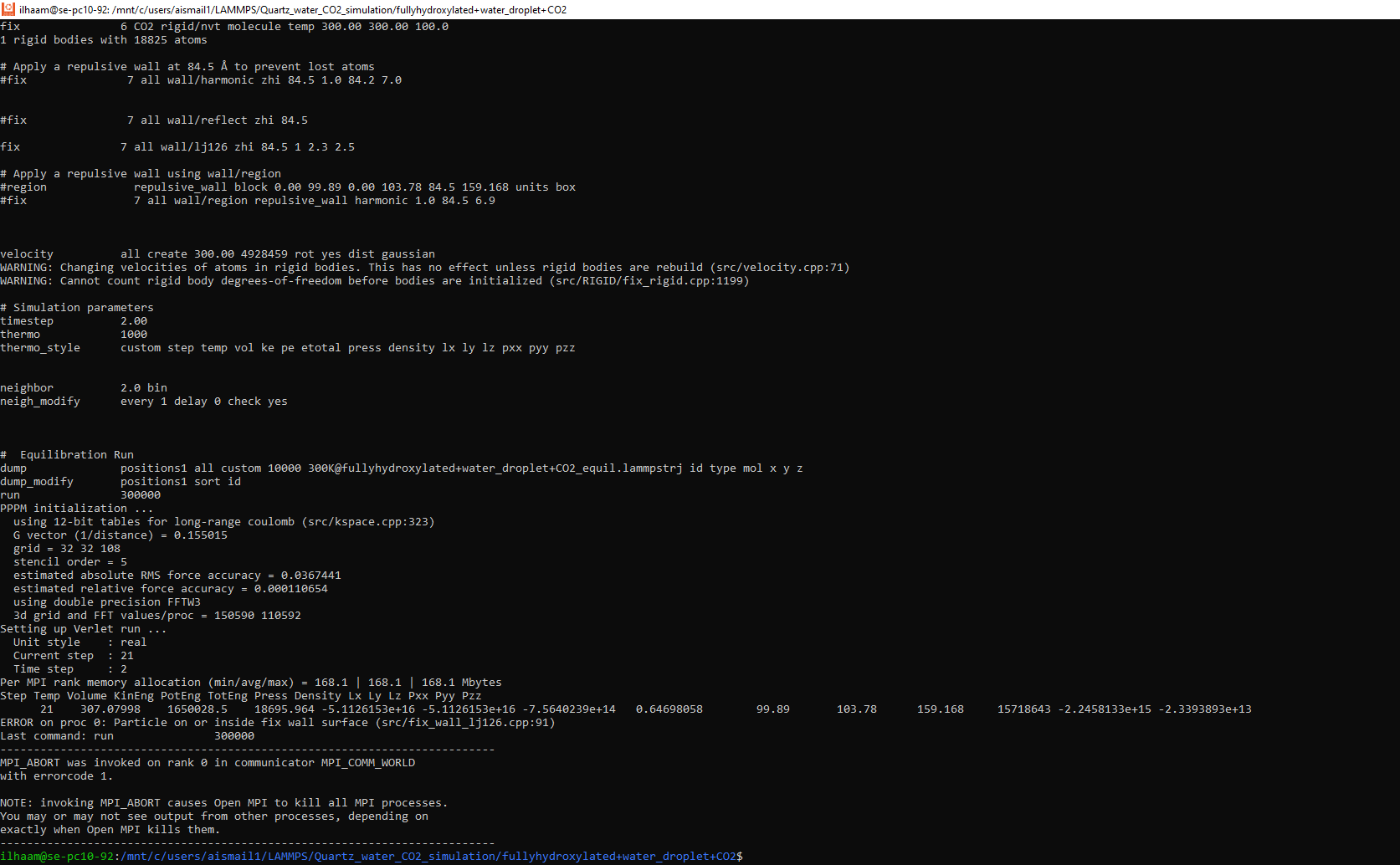
I am using LAMMPS 22, and I created a system of hydroxylated quartz with water droplet and CO2 molecules on top of it, the simulation is performed with periodic boundary conditions in x- and y-directions. In the z-direction, a repulsive force is set at 60 A above the tip level of OH (24.5A). I have tried wall/lj126, wall/harmonic, and wall/region. I keep getting the error below for all the wall commands.
ERROR on proc 0: Particle on or inside fix wall surface
I was going to suggest these changes for the wall issue:
processors * * 1 # better for slabs.
pair_style lj/cut/coul/long 12. # 17 is an overkill
minimize 0.1 0.1 1000 10000 # let's be quick here
# Use walls to prevent lost atoms
fix 7 all wall/reflect zhi EDGE
fix w2 all wall/lj126 zlo EDGE 1.5 2.3 6. # change zlo to -2.8 in the DATA file.
timestep 1.00 # 2 was definitely too optimistic.
but then noticed a far more serious issue: you have only 3 molecule IDs in the system: one for quartz, one for all water molecules, and one for CO2. That’s not how a type-I force field works. For SiO2 I believe you have to assign every atom to a different molecule, while for CO2 and water, one ID per molecule will do
Thank you very much for your response. I did consider suggestion and edited the data file to include all the numbering for the fluid molecules. and with your suggestion with the wall, ALSO from LAMMPS documentation it is mentioned not to use fix wall/reflect with fix rigid command. with the CO2 i used FIX RIGID for the temperature. fullyhydroxylated+water_droplet+CO2_editted_data.data (2.3 MB)
did input everything you recommended and i had the Error below. attached again is the modified data file. Thank you
ERROR on proc 0: Out of range atoms - cannot compute PPPM (src/KSPACE/pppm.cpp:1936)
This is not the way to proceed. When you have lost atoms, it means either:
A bad physical set-up.
Bogus input.
Inconsistent combination of LAMMPS commands.
A combination of the above.
Now, your input data file shows that:
The SiO2 slab is still assigned to a single molecule ID. Also, you defined a bond between O–H for the surface hydroxyl groups, but not one between Si–O, even though there is an angular term involving the three atoms. See for instance the selection index 1798 7558 17488 in VMD.
The same molecule ID is reused for different species, e.g. resid 2 shows a water and carbon dioxide molecule.
You are right about not using a reflective wall to contain rigid molecules. The bottom line is that this is a rather complex simulation and you have to do some effort to debug it. I would start by simulating each piece separately, e.g. begin by verifying that your setup for the quartz slab is correct, then add the water droplet, and finally the carbon dioxide. I suspect that the surface is the offender here, but I have no desire of going further with my analysis.