Following up on a previous thread (Regions NOT forming single region in region union command - #14 by mmoosa92), I first intended to create a “complex” wall using the union of sub-regions. This region comprises of a cylinder connecting two boxes, similar to the one discussed in the linked thread above. The goal was to use fix wall/region to apply a “soft” repulsion of the atoms and keep them contained in the region. However, it was advisable that such complex region can suffer from some ambiguities, especially when an atom crosses the planes between the sub-regions, and this is exactly what I observed in my simulations. Hence, I am creating the wall with explicit atoms with the following commands:
lattice bcc 1
region simbox block -50 50 -50 50 -55 64.6 units box
create_box 3 simbox
region pore cylinder z 0.0 0.0 11.1 0 14 units box
region boxOne block INF INF INF INF -50.6 0.5
region boxTwo block INF INF INF INF 13 EDGE
region freeSpace union 3 pore boxOne boxTwo
region carveBoxOne block INF INF INF INF -49.9 -0.1
region carveBoxTwo block INF INF INF INF 13.6 63.6
region carvePore cylinder z 0 0 10.1 -0.1 13.5 units box
create_atoms 3 region freeSpace
delete_atoms region carveBoxOne
delete_atoms region carveBoxTwo
delete_atoms region carvePore
With the above commands, I get the following walls (my simulation box is periodic in x and y, that is why I “carved” the atoms all the way along in those directions):
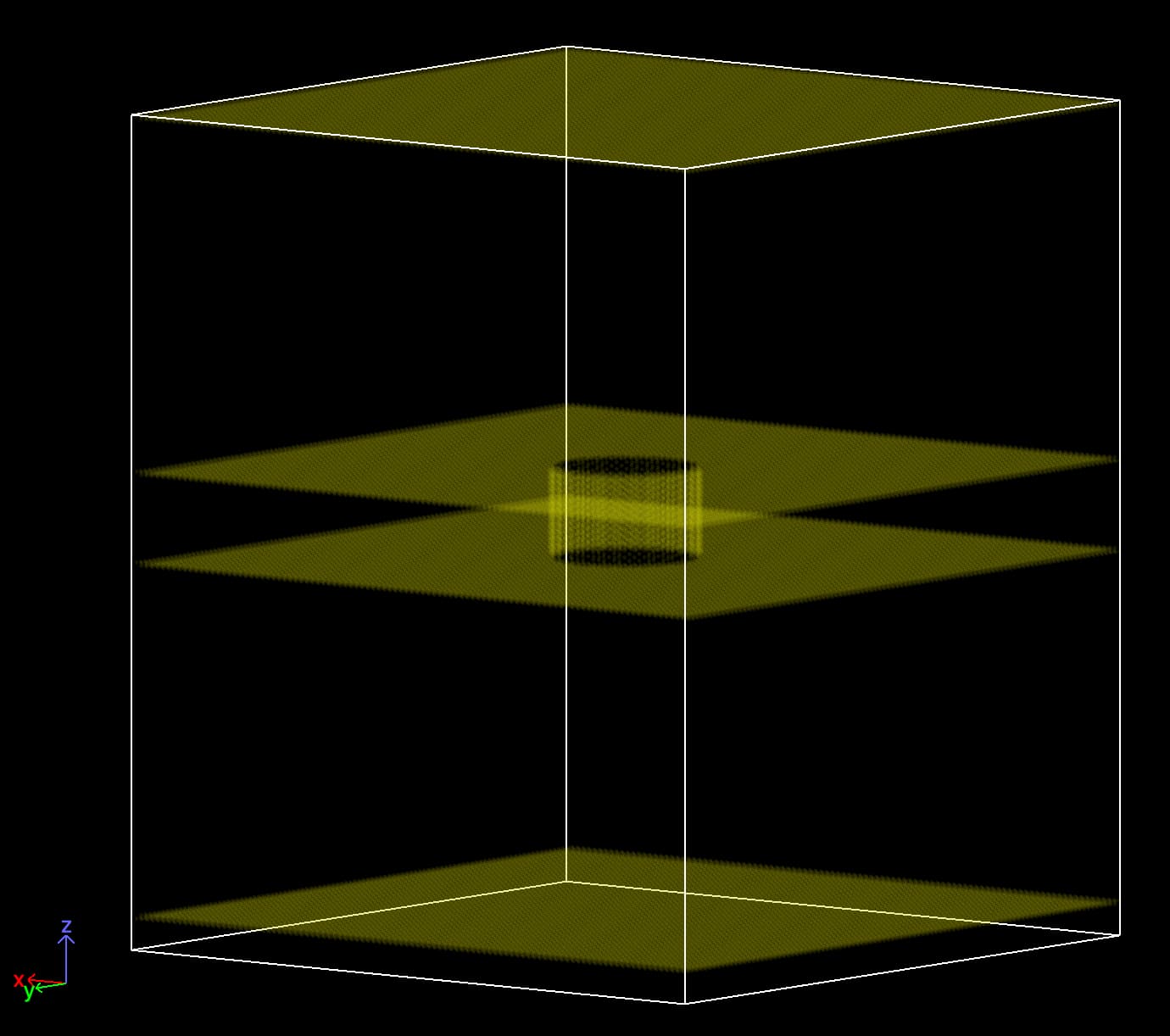
where I made all the walls double layered (and staggered using the BCC lattice). Below is a side view of the cylindrical region showing the side walls with two layers.

Just to test the integrity of this wall, I simulated some atoms (starting from one box) through Langevin Dynamics and NVE integrator. I am also adding a force from a table through fix wall/table simulating the effect of the solvent and pore implicitly. I am facing two issues:
(1) By simulating the wall using explicit atoms, the computational time increases significantly*. I used fix setforce to set all the forces to zero for the wall atoms. Is there a more efficient way to do this without compromising the computational efficiency significantly? When using multiple processors, I used the following commands:
neighbor 2.0 bin
neigh_modify exclude type 3 3
where type 3 is for the wall atoms. This speeds up the calculations. However, I want to know what are some suggestions to maximize the computational efficiency even when using a single processor.
* I have less than a 100 mobile atoms. For 1 ns, simulation time increases from a couple of seconds to a couple of hours!
(2) Even with a double-layered wall as described above, my simulation often fails due to an atom penetrating the wall (specifically the bottom wall). I am using lj96/cut 1.0 1.0 for the interactions between the wall atoms and all other atoms in my simulation box. Perhaps increasing the \sigma will result in a more packed wall and that can prevent any penetration, however that did not work (for \sigma up to 3.5). Is it advisable to make the wall thicker? However, in doing so, I would have more atoms and this brings me back to the first inquiry. Also, is there a better pair style to use instead of lj96? I intended to use lj93 but it is not available as a pair style but rather in fix wall. I really just want to “softly” bounce the atoms of the walls (simulation box edges and the pore wall). I used the regular lj/cut pair style and I do not see any penetration for at least 10 ns of simulation time, but I was seeking to implement a “softer” repulsion. Any suggestions?
PS: I am using LAMMPS 02Aug2023 version.
Any input is appreciated. Thanks!