Hi Axel,
Thank you very much for all the insight and the indication of what to correct.
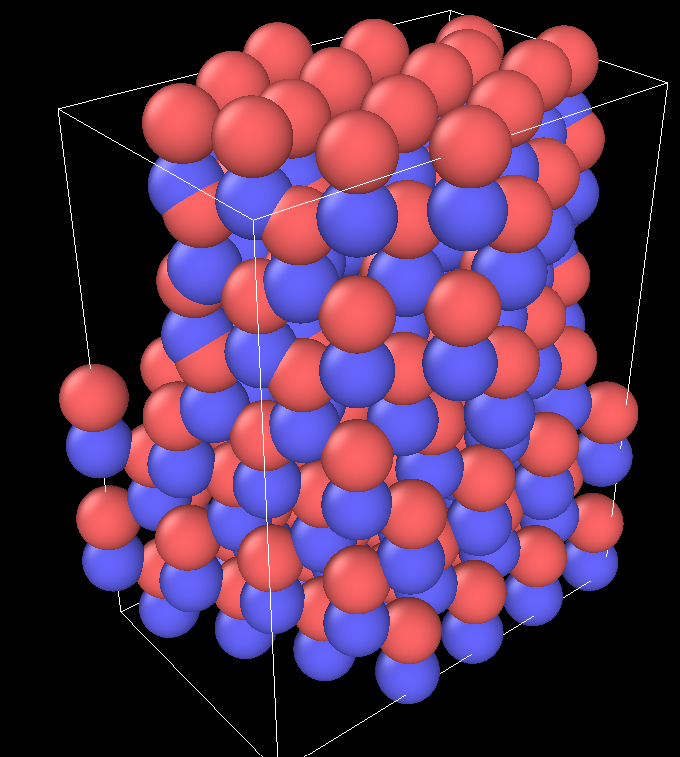
I did prefer to import a crystalline structure (read_data) instead of the lattice creation because I had some previous issues with this data you mentioned. It worked to read and join 2 ZnO crystals on the planes (0001) and (000-1) with a twist of 27.8° to create the grain boundary (GB). I’m also determining an outside region and group to delete and leave an orthorhombic assembly of 2 crystals.
Further I’ll need to use lx and ly of the box to calculate the GB area, but these remain with the value of the 2 triclinic boxes assembled. I’ve tried to use change_box to reestablish the box dimensions to the orthorhombic, but the follow error appears:
ERROR: Triclinic box skew is too large (…/domain.cpp:216)
Last command: change_box upper x final 5.18 26.18 boundary p p p set units box
Do you have any suggestions on how to change this box dimensions?
Thank you in advance!
Best,
Gustavo
Part of the script:
— Create Atoms
read_data ZnO_lower.xyz
region lower block INF INF INF INF INF INF
read_data ZnO_upper.xyz add append offset 0 0 0 0 0 shift 0.0 0.0 106.13642
region upper block INF INF INF INF INF INF
region outside block 5.18 26.18 22.72 49.89 0.00 212.28 side out
group upper region upper
group lower region lower
group outside region outside
change_box upper x final 5.18 26.18 boundary p p p set units box
#change_box lower x final 5.18 26.18 boundary p p p set units box
#change_box upper y final 22.72 49.89 boundary p p p set units box
#change_box lower y final 22.72 49.89 boundary p p p set units box
— Force Field
pair_style reax/c lmp_control
pair_coeff * * ReaxFF_Potential_ZnOpure O Zn
neighbor 2 bin
neigh_modify every 10 delay 0 check no
fix 1 all qeq/reax 1 0.0 10.0 1e-6 param.qeq
---------- Displace atoms and delete overlapping atoms ---------------------
#displace_atoms upper move 0 0 0 units box
delete_atoms overlap 1.0 lower upper
delete_atoms group outside
— Define Settings
compute csym all centro/atom 12
compute eng all pe/atom
compute eatoms all reduce sum c_eng
---------- Run Minimization ---------------------
…
in.GB_S13_0001_tw_GBE.ZnO (3.25 KB)