Hi,
I’m trying to make Lammps data file for an ionic liquid. I’m using the force field information from the SI of this paper (https://doi.org/10.1016/j.jil.2022.100040).
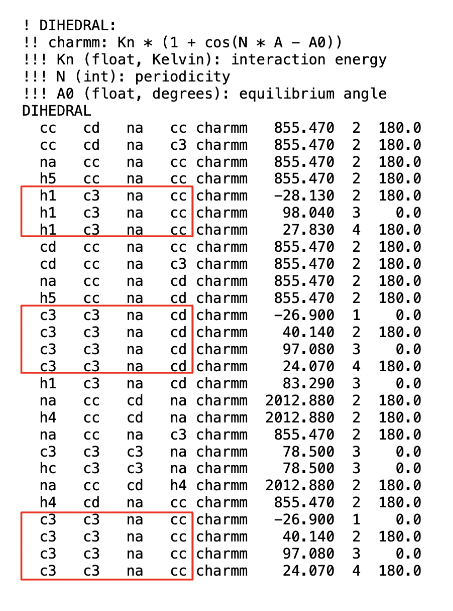
While writing the Dihedral table, I need to assign a dihedral type to them. I have attached a screenshot of the Dihedral coefficients from SI where some of the dihedral types look redundant but have different coefficients. Do I assign these coefficients to a unique dihedral type? If so, how do I identify which dihedral is of a certain type given that all the four atom types are matching?
The paper says they use the GAFF functional form, which sums the equation Kn * (1 + cos( N * A - A0) ) over the listed coefficients (which I believe is the ‘fourier’ implementation in LAMMPS ). If they are using GAFF, then coefficients listed for the same dihedral atoms won’t become unique dihedral types, but will instead contribute to a single equation
SUM( Kn * (1 + cos( N * A - A0) ) )
1 2 3 4 Kn N A0
h1 c3 na cc charmm -28.130 2 180.0
h1 c3 na cc charmm 98.040 3 0.0
h1 c3 na cc charmm 27.830 4 180.0
so that the total equation for the dihedral energy becomes
Thanks for the response!
So instead of using the dihedral_style charm, I should use the dihedral_style fourier and combine the coefficients for dihedrals of the same atom types in a single dihedral type?
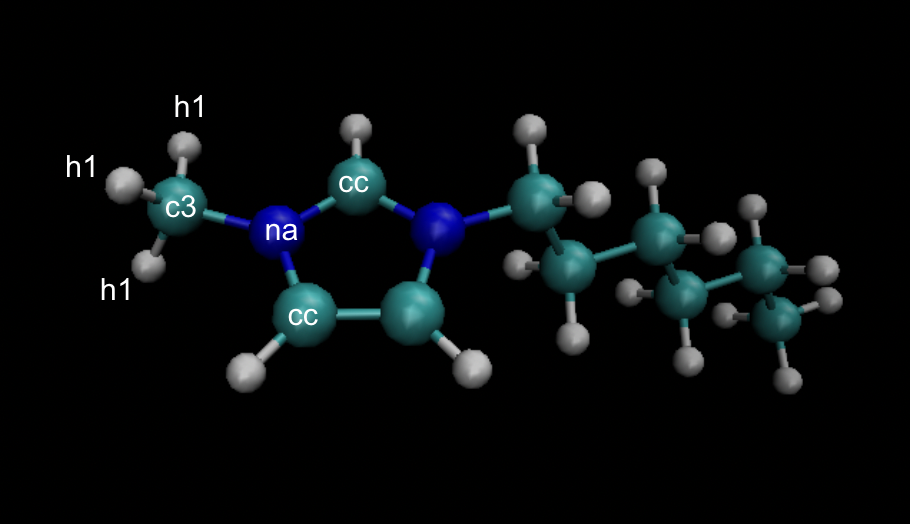
Also, it seems like there are multiple dihedrals with atoms “h1 c3 na cc” in the molecules. So while writing the dihedral table in the datafile ("n Dihedral_type Atom1 Atom2 Atom3 Atom4), should I give all these dihedrals the same dihedral type?
Yep, exactly on both. There will be a single fourier entry for that dihedral type with all the coefficients, and for the atoms labeled, it looks like there should be 6 dihedrals entries, all of the same cc-na-c3-h1 (or h1-c3-na-cc) type