I am attempting to simulate a dilute solution of plate particles using Langevin dynamics. The model for the plate particles runs fine, however, I am experiencing an issue when measuring the normal stresses in equilibrium.
After inspecting the source code, I believe this is due to create_atoms random using the geometric centre of the molecule for the random distribution of the plates, however, it would be more appropriate to randomly distribute the normal vector to the plate to guarantee there is no bias to certain orientations.
Can anyone suggest a way to achieve this via lammps commands?
Of course, I could produce the particle positions myself to verify the distribution of the normal vector, then I would produce a data file for this.
What is a “plate particle”?
There is no mention of this type of particle anywhere in the LAMMPS manual.
But that is what it is supposed to do!
That is supposed to happen, if you create a molecule instead of an atom. Molecule files may have only one particle…
Please note that your message is “encoded with the specifics of your research” and thus hardly comprehensible for anybody that is not familiar with your research, i.e. pretty much anybody reading this. Furthermore, it is very difficult to discuss on the basis of a rather unspecific description that does not show the exact details of the sections of source code you are referring to and does not provide any details or examples of how what you see can be reproduced.
Bottom line, if you want to have competent and specific help, you have to assume that people here no nothing about anything you are doing and thus explain everything like to a stranger. The forum guidelines discuss this at length. Perhaps you need another careful read of them.
This is just 6 particles connected via harmonic springs and angles to form a triangular “plate” structure. This information is held in the molecule file I created.
“But that is what it is supposed to do!”
Yes, but In this case it does not account for the orientable structure of the plate. Hence why I am asking for advice.
I am trying to ask a specific, separate question relating to how create_atoms would produce random orientations rather than the random distribution of centroids that I currently obtain.
“Furthermore, it is very difficult to discuss on the basis of a rather unspecific description that does not show the exact details of the sections of source code you are referring to and does not provide any details or examples of how what you see can be reproduced.”
I did not think that would be needed given the nature of my question, I am not suggesting Lammps is doing something incorrectly, nor am I experiencing a bug. I am looking for a DIY approach to solving the problem.
Furthermore, my results have come from thousands of realisations which would be far too much effort for someone to reproduce.
I will try and make the question more clear,
Given a planar triangular molecule, made from 6 particles, 3 at each vertex and 3 in the midpoints, is there a way I can randomly distribute the normal vector of this object ? The normal is the cross product of 2 of then vectors which describe the sides of the triangle. create atoms does not seem to achieve this goal.
So why don’t you just say that you are trying to create “plate shaped molecules” from molecule files. This is precise and describing what you are doing within the framework of what LAMMPS knows about (and thus other LAMMPS users and developers). By using your research specific “slang” you are just excluding people that could provide advice by them looking at your post and concluding that they know nothing about this, even though they might. I am not trying to waste your time, but trying to show you how to get more and better help.
But that is supposed to happen when you use molecule files. From the documentation:
This is done by placing the geometric center of the molecule at the lattice point, and (by default) giving the molecule a random orientation about the point. The random seed specified with the mol keyword is used for this operation
Again, you are making it very laborious to give a meaningful answer. If you had instead produced a minimal input deck where you create a small number of such particles and demonstrate what you are trying to achieve, it is much less work to try and understand what you are looking for and reproduce and possibly suggest changes. By just giving a (vague) description, people will have to go through the extra step to create such an input and may possibly do it differently and then do not experience what you see.
You don’t need to report thousands, one of a few representative ones would suffice.
You are not understanding how a forum works and how people can help you. When asking non-specific questions without examples, you are implicitly assuming that everybody will do exactly the same thing as you do based on your description. That is rarely the case. Also, you are forcing the people trying to respond to do more work. The rule for getting help is very simple: the more effort it is to respond, the smaller the chance to get help.
Thus, by using a general description instead of a specific one with a reproducible example, you are restricting the range of people that could respond and force them to do more work, which also restricts the number of people. So it is in your own best interest to always do what I suggest.
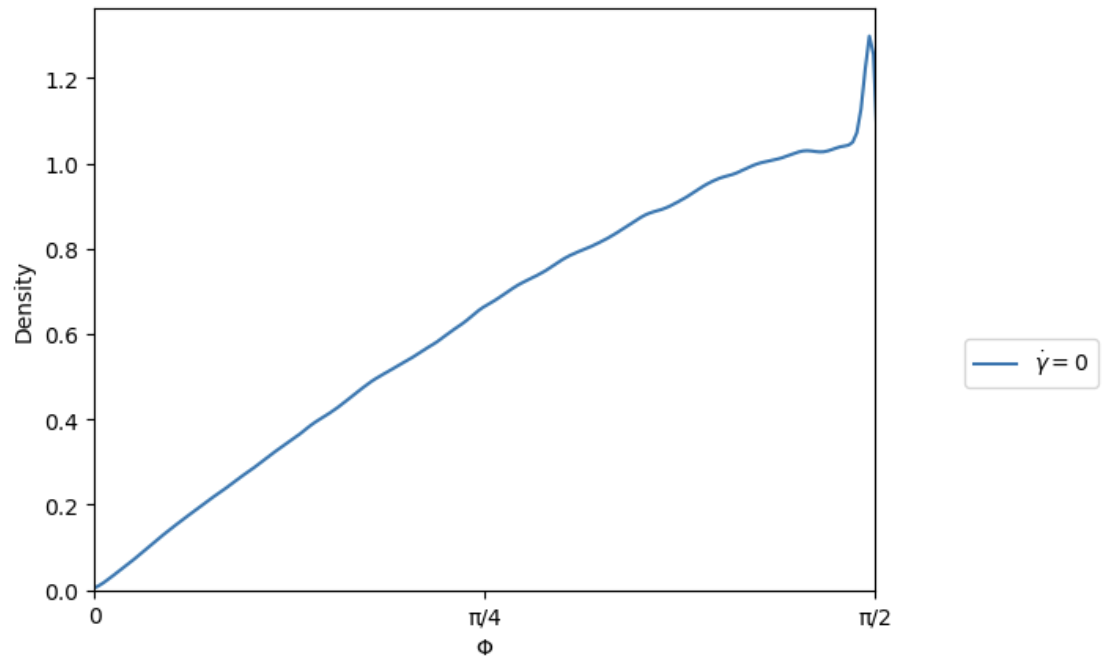
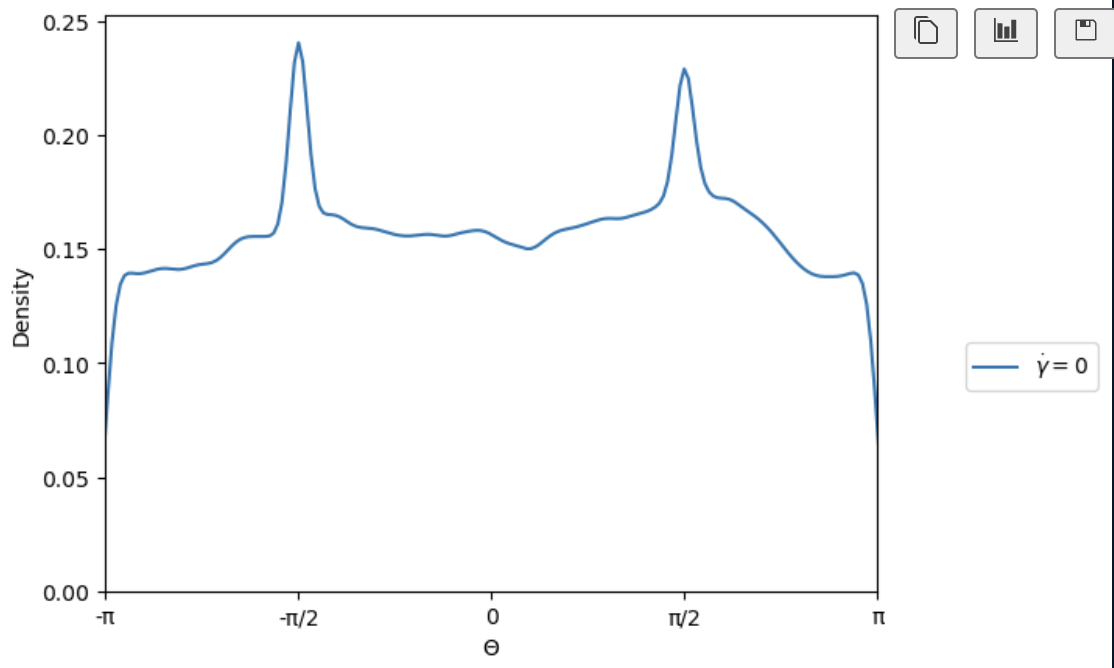
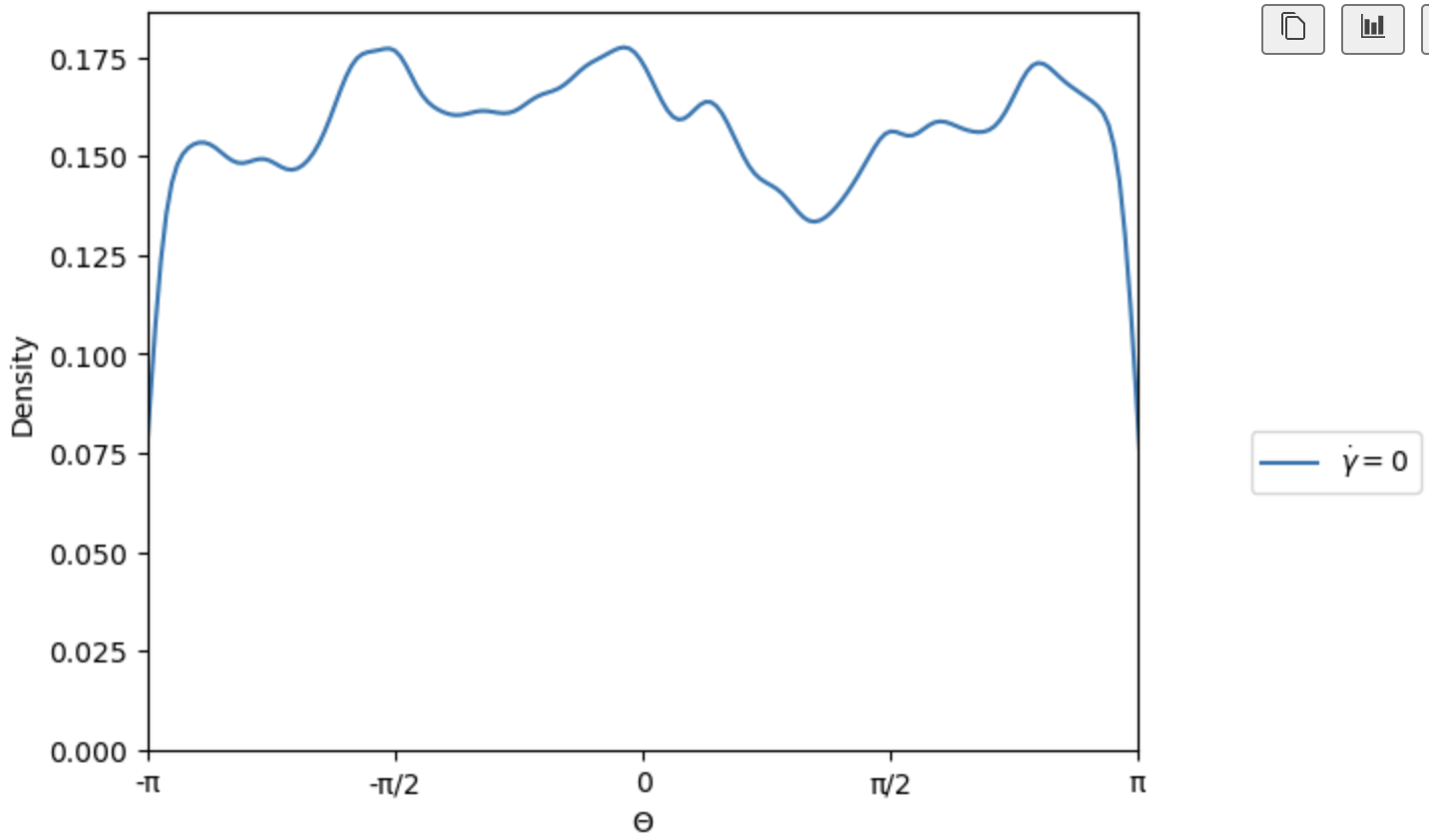
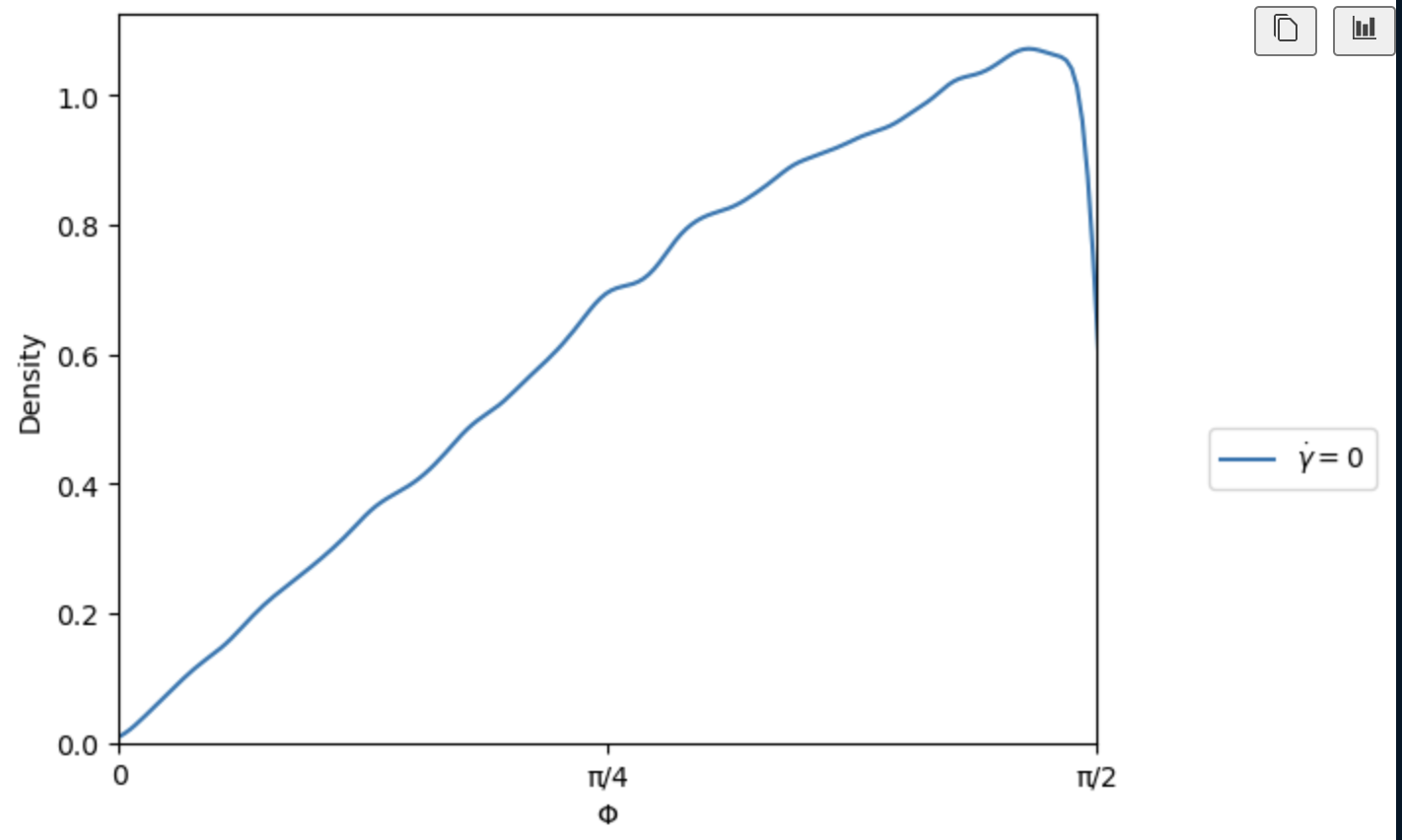
I understand, however when I run my simulation and calculate the orientations of the plates, I get the following distributions for theta(azimuth) and phi(inclination). I have shifted all orientations to sit in the upper hemisphere.
At this point, I can equilibrate the simulation before applying my driving forces which will have the desired orientational distributions.
I am still interested to understand if there is a way to improve my script so that the distribution is achieved upon insertion of the particles with create_atoms.
Very obviously the different “molecules” have different orientations. Those are random, but not necessarily following a specific distribution. I don’t think that makes much sense with placing them also at random positions. You obviously must spend sufficient simulation time on equilibrating this.
Issues with pressure, assuming it is caused by particles being too close, can be reduced when using the overlap keyword of create_atoms random. I don’t think that there is anything else that can be done on the LAMMPS side. I cannot comment on the rest, since this is research too far away from from stuff where I have some experience.