Hi, all
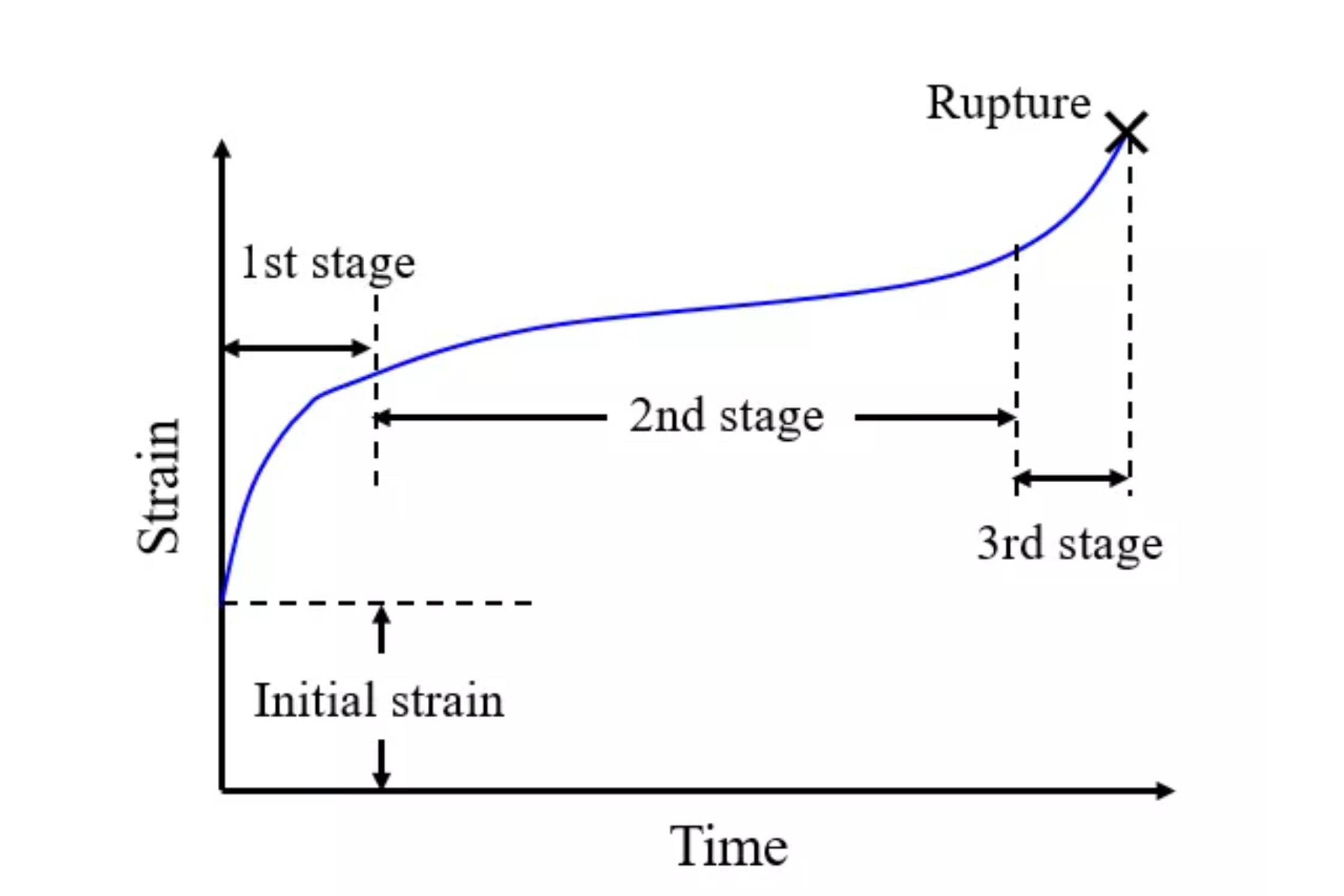
I am trying to simulate a creep test for a high entropy superalloy and draw a diagram with time vs. strain
Below is my input script
I omit the part of building the model in the script
#----------initial------------
velocity all create 1033.0 4846515
variable TimeStep equal 0.001
variable Time equal ${TimeStep}*step
variable T equal 1033.0
variable Pressure equal 0.0
variable TDamp equal ${TimeStep}*100
variable PDamp equal ${TimeStep}*1000
#------ running ------
reset_timestep 0
timestep ${TimeStep}
#------ stage 2 ------ run npt thermostat
#assign a random velocity to atoms at 1033K
region left prism 0.0 5 INF INF INF INF 0.0 0.0 0.0 units box
region right prism 163.16 168.16 INF INF INF INF 0.0 0.0 0.0 units box
group left region left
group right region right
group mobile subtract all left
compute 5 mobile temp
velocity mobile create ${T} 4928459
velocity left create 0.0 4928459
fix freeze left setforce 0.0 0.0 0.0
fix eq3 all npt temp ${T} ${T} ${TDamp} aniso 1.01325 1.01325 ${PDamp}
fix_modify eq3 temp 5
thermo 1000
thermo_style custom step temp c_5
run 6000
unfix eq3
#------ running ------
reset_timestep 0
timestep ${TimeStep}
#-------prepare---------------------------------
variable tmp equal lx
variable L0 equal ${tmp} #initial length
compute 3 right displace/atom #delta x(vector)
compute strainn right reduce ave c_3[4] #translate into scalar
change_box all boundary f p p
change_box all x delta -150 150 units box
#------ stage 3 ------ Store final cell length for strain calculations
variable maxforce equal 0.1
variable minforce equal 0.04
variable totalrun equal 200000
variable force equal ${minforce}+((${maxforce}-${minforce})*step/${totalrun})
fix n1 all npt temp ${T} ${T} ${TDamp} y 1.01325 1.01325 ${PDamp} z 1.01325 1.01325 ${PDamp}
fix_modify n1 temp 5
fix d2 right addforce ${force} 0.0 0.0
variable strain equal c_strainn/v_L0 #strain
restart 40000 *_.restart
dump d1 all custom 1000 strain_*.dump id type xu yu zu
dump_modify d1 sort id
thermo 1000
thermo_style custom step press temp c_5 v_L0 v_strain
run ${totalrun}
unfix n1
unfix d2
#unfix d2
unfix p1
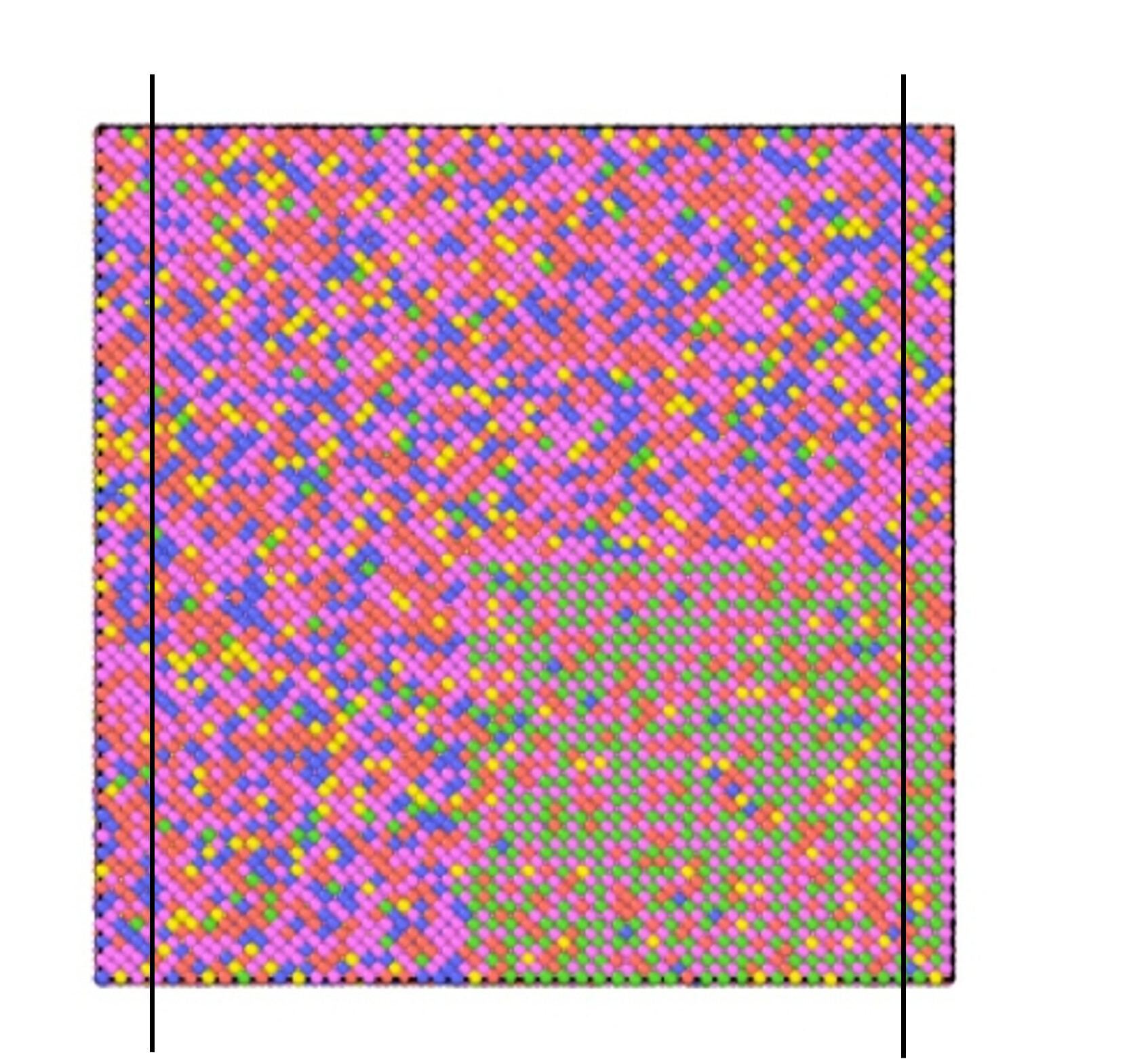
the boundary is f p p
I divided the model into three parts as the picture above
I let the left part fix setforce 0.0 0.0 0.0
and the right part fix addforce in x direction
and get the video below
it should keep stretching rather than oscillating
how could I do to fix this
if there is any information I didn’t offer, please tell me