Hello,
I am currently trying to create the lammps data file for protein using below command
perl charmm2lammps.pl HAP 1qz1 -charmm
it is able to convert into data file, but some of the bond parameters, dihedral, angle parameters are missing.
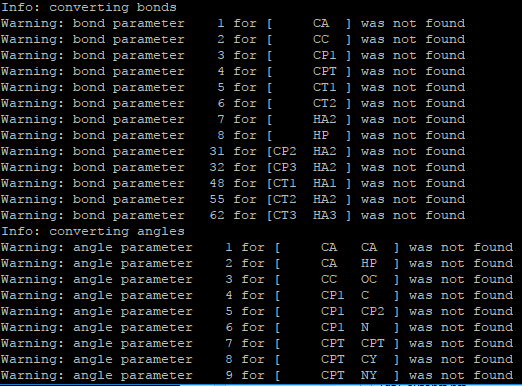
can you suggest how to solve this problem. I am using charmm22 Topology File for Proteins.
Hello,
I am currently trying to create the lammps data file for protein using
below command
perl charmm2lammps.pl HAP 1qz1 -charmm
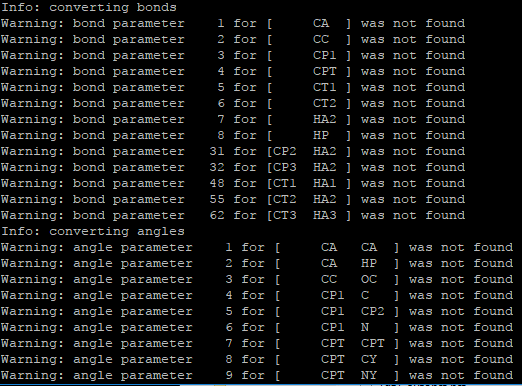
it is able to convert into data file, but some of the bond parameters,
dihedral, angle parameters are missing.
!image.png|522x386
can you suggest how to solve this problem. I am using charmm22 Topology
File for Proteins.
first off, CHARMM 22 is *way* outdated.
there is not much information to go on here. how did you obtain the .psf
file?
did you test your coordinate/topology files with a program that can read
CHARMM topologies natively (e.g. CHARMM or NAMD)?
which version of the charmm2lammps.pl script are you using?
axel.
Hello,
Thank you for the reply,
1. I have tried using top_all36_prot (CHARMM36 All-Hydrogen Topology File
for Proteins), it resulted less benefit in conversion than using charmm
22. That's why I used the charmm22.
you should use the same version or newer, that was used to create the psf
file. for simulating proteins, you should also include cmap corrections.
2. Can you suggest where to get the right updated topo charmm file ?
the canonical location for charmm force field parameters is here:
http://mackerell.umaryland.edu/charmm_ff.shtml#charmm
3. I have generated the psf using autopsf generation tool using vmd.
then you have to be very careful. autopsf simplifies generating topologies
by automating tedious tasks, but if you have any unusual components in your
system, it may create bogus files.
4. I am currently using charmm2lammps.pl version is 2005.
the current version of charmm2lammps.pl identifies itself as version 1.9.1
with changes from 2016
5. I have checked the pdb and psf generated in the vmd.
that is not sufficient to determine if the topology build was correct. you
cannot check for proper atom type assignment or
bond/angle/dihedral/improper type.
axel.
Hello,
Thank you for your suggestion,
Now I am able to create the lammps data file completely.
I have been using the different topo file in autopsf generation.
As you suggested when I used with topo file along cmap correction, it had assigned all the properties correctly in psf file.
Regards
Mohan
{kind=link}