Hi, after obtaining the polymer model from moltemplate, I used fix deform to reduce the size of the box from (300, 180, 180) to (180 180 180). Following that, I hope to do NVT.
The protocol is to create a well-mixed polymer system (aka like a melt) for further simulation.
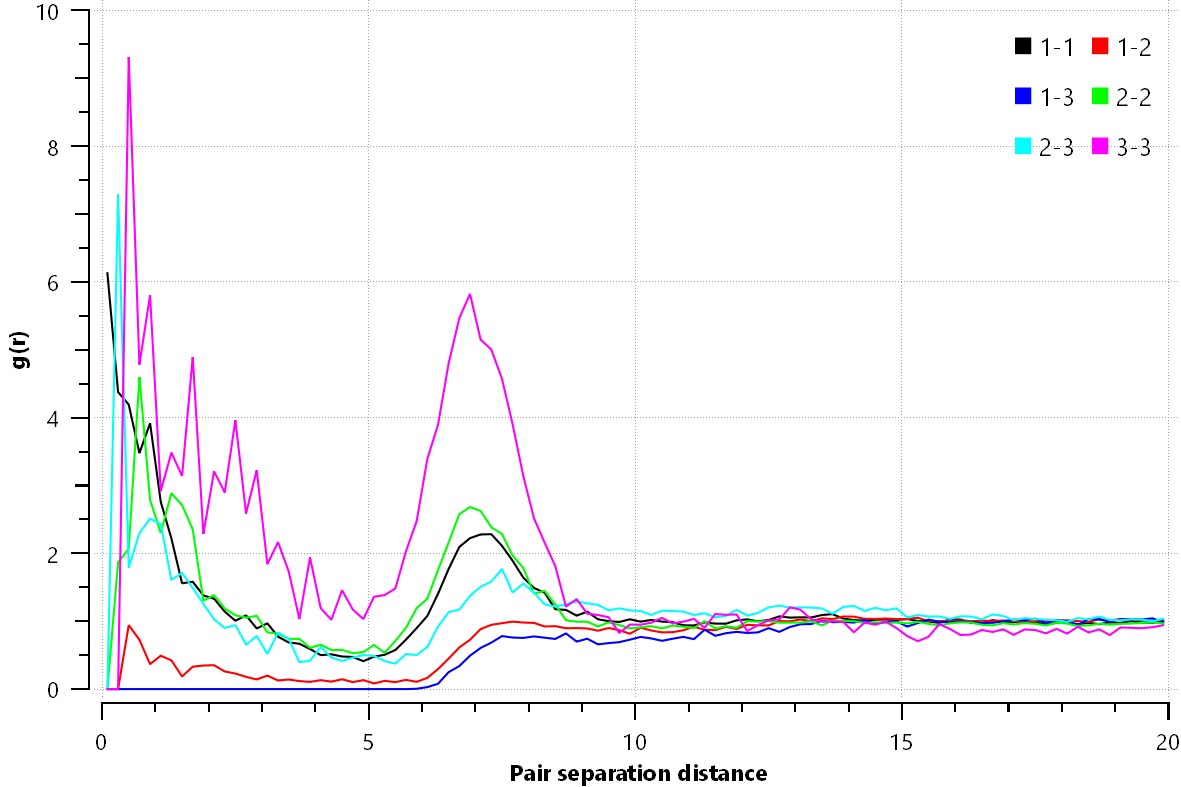
The problem is that my rdf, after 5,000,000 steps, looks like the one I uploaded.
units real
atom_style full
bond_style harmonic
pair_style lj/cut 14
read_data ${to_read_data}
bond_coeff * 0.6 7
pair_coeff * * 0.148 7 14
timestep 2.0
thermo_style custom step temp pe etotal press vol density lx ly lz
thermo 5000
fix 2 all deform 1 x final 0 ${lx}
run 50000
unfix 2
write_data system_after_deform_lx-${lx}.data
##########
# NVT ensemble - 1
thermo_style custom step temp pe etotal press vol density lx ly lz
thermo 5000 # time interval for printing out "thermo" data
dump 1 all custom 10000 nvt-1.dump id mol type x y z ix iy iz
fix 1 all nvt temp 300.0 300.0 $(100.0*dt)
restart 5000 nvt-1.restart
run 5000000
unfix 1
write_data nvt-1.data
Any suggestions on how to proceed?