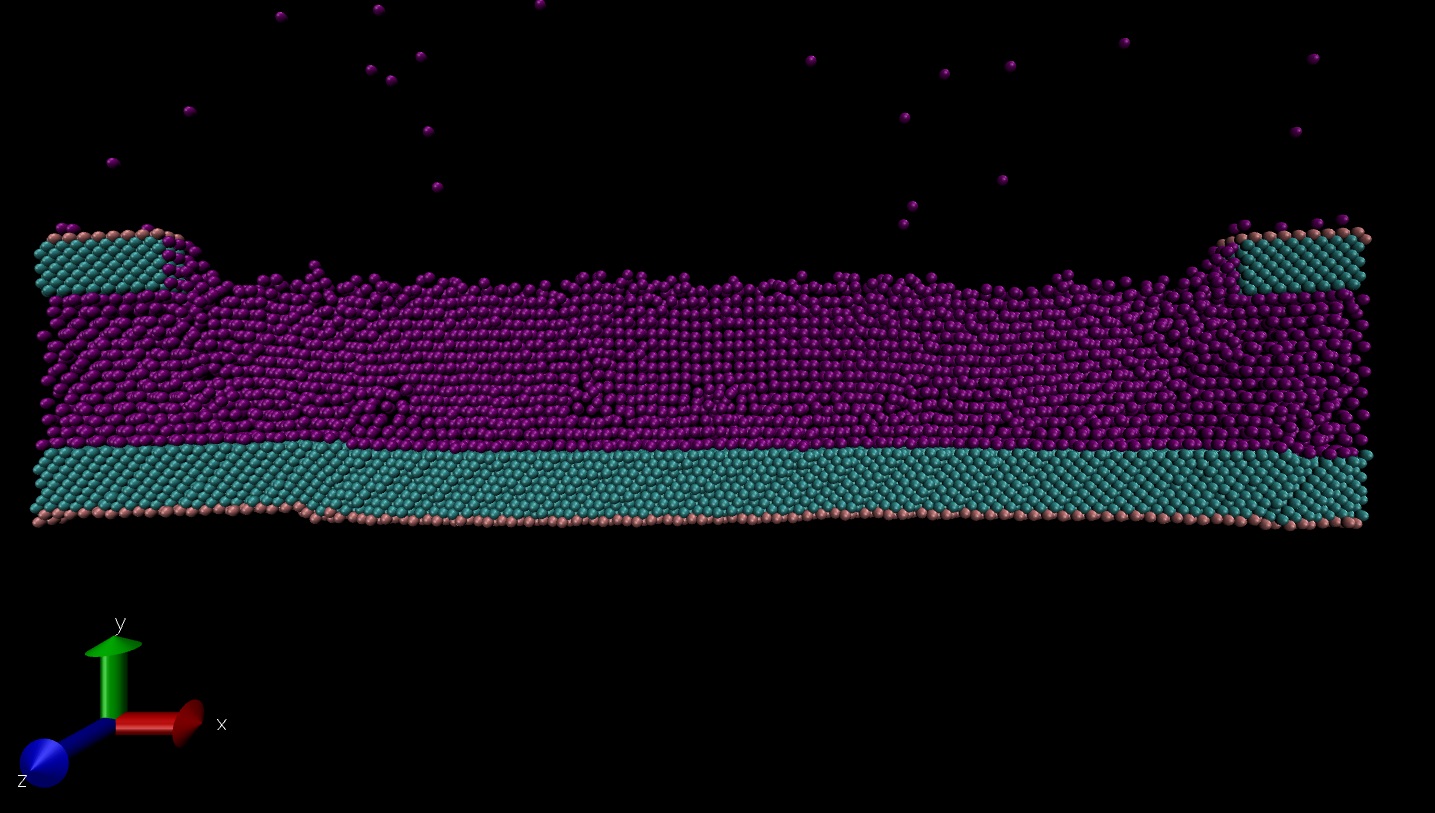
I am trying to simulate an evaporating meniscus formed by liquid argon between two parallel copper walls, with an opening in the upper wall. After the equilibrium stage, there is deformation in the copper walls as you can see in the attached image.
If you want to stop the copper atoms from moving and/or keep the copper wall rigid, there are various approaches that have been discussed numerous times.
Regarding people from the mailing list reviewing your input line by line, you may receive a message from Axel shortly.
Hi Giacomo. the innermost and outermost layers of copper are “fixed” layers using the following commands:
velocity wall_fix set 0.0 0.0 0.0 units box
fix 1 wall_fix setforce 0.0 0.0 0.0
But even those fixed copper layers are still deforming!
Thank you!
If you want to stop the copper atoms from moving and/or keep the copper wall rigid, there are various approaches that have been discussed numerous times.
Regarding people from the mailing list reviewing your input line by line, you may receive a message from Axel shortly.
I am trying to simulate an evaporating meniscus formed by liquid argon between
two parallel copper walls, with an opening in the upper wall. After the
equilibrium stage, there is deformation in the copper walls as you can see
in the attached image.
[...]
I would really appreciate if you could guide me how to resolve this issue.
you can resolved this through a process called "debugging". that is
something, that *everybody* has to do to figure out problems they created
themselves in their input.
for example, start a new, empty input file and copy over the input from the
existing input piece by piece but build as small a system as possible and
leave out all parts that are not immediately requred, so that you build
your system in steps, while you validate at each step, that all properties
and assumptions are as you expect them.
you can use the info command to do introspection of many LAMMPS properties,
also, you can check overlap of group membership easily through the group
intersect command, also you can write out dump or data files to check all
properties and coordinates and velocities. and so on and so forth.
people here will typically look at input files and get back to you in two
cases: 1) there is a glaring mistake, that immediately jumps off the
screen, 2) there is a convincing indication, that there may be a bug. posts
to the mailing list of the kind "please tell me how to fix my input" have a
rather low threshold to be looked at seriously.
Thank you very much for your time and detailed explanations.
Best,
Santiago
Dear LAMMPS users,
I am trying to simulate an evaporating meniscus formed by liquid argon between two parallel copper walls, with an opening in the upper wall. After the equilibrium stage, there is deformation in the copper walls as you can see in the attached image.
[…]
I would really appreciate if you could guide me how to resolve this issue.
you can resolved this through a process called “debugging”. that is something, that everybody has to do to figure out problems they created themselves in their input.
for example, start a new, empty input file and copy over the input from the existing input piece by piece but build as small a system as possible and leave out all parts that are not immediately requred, so that you build your system in steps, while you validate at each step, that all properties and assumptions are as you expect them.
you can use the info command to do introspection of many LAMMPS properties, also, you can check overlap of group membership easily through the group intersect command, also you can write out dump or data files to check all properties and coordinates and velocities. and so on and so forth.
people here will typically look at input files and get back to you in two cases: 1) there is a glaring mistake, that immediately jumps off the screen, 2) there is a convincing indication, that there may be a bug. posts to the mailing list of the kind “please tell me how to fix my input” have a rather low threshold to be looked at seriously.