Hi All,
I’m seeing a inconsistent behavior when I use “delete_bonds”. I posed a question regarding this issue while I was working on load balancing of a simulation which is available in:
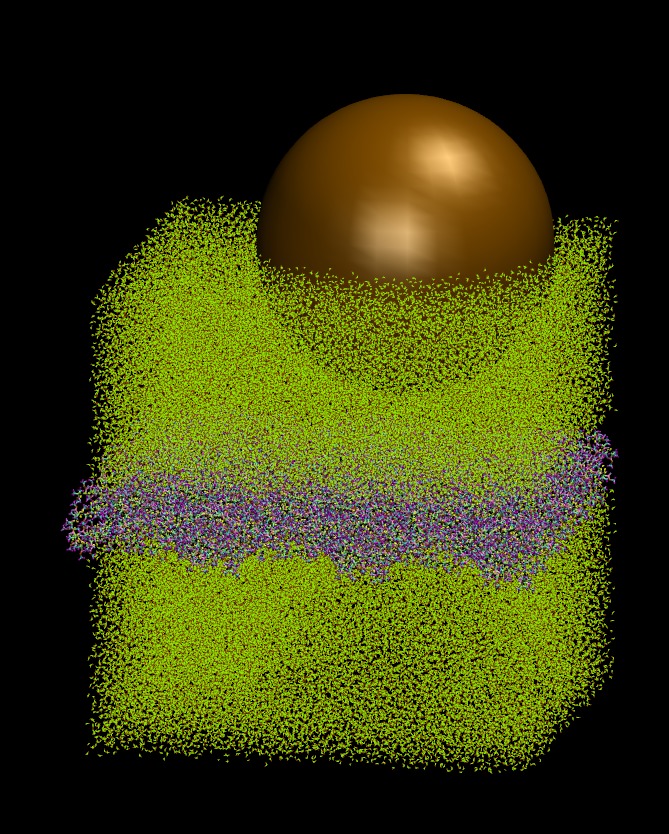
The problem is the same one that I described in above thread, i.e. a big colloidal solute (10nm in diameter) is in water which is covering both sides of a solid polymeric wall (see attached figure). In order to keep the wall in place, I define a block within the wall (with a width of 5anstrom and its xy plane extent are the same as the wall itself) and I exclude this block from integration and I also delete the bonds within this block to eliminate their potential contributions to the simulation. However, deleting bonds is where I see some peculiar behavior which is not identically reproducible each time that I run the system! If there is no colloidal particle in the system and I’m not setting neighbor lists and cut offs to satisfy the colloidal interaction requirements, delete_bonds works just normal. However, in the presence of the colloidal particle each time that I run the simulation I see following the behaviors sporadically: – Sometimes it smoothly pass the “delete_bonds” and the simulation goes as it should – Sometimes it gets stuck forever in “Deleting bonds …” mode as I reported in (). – Sometimes it aborts the job by giving the following errors: == When I was using LAMMPS (11 Sep 2015-ICMS) as I reported in () : : : Deleting bonds … [cli_79]: aborting job: application called MPI_Abort(MPI_COMM_WORLD, 1) - process 79 == Now I’m using LAMMPS (13 Jan 2016-ICMS) and at times that I get errors they’re either of the following ones: ** Deleting bonds … ERROR on proc 139: Failed to reallocate 1310720 bytes for array atom:bond_atom (…/memory.cpp:66) [cli_139]: aborting job: application called MPI_Abort(MPI_COMM_WORLD, 1) - process 139 OR ** Deleting bonds … ERROR on proc 29: Failed to reallocate 1572864 bytes for array atom:nspecial (…/memory.cpp:66) [cli_29]: aborting job: application called MPI_Abort(MPI_COMM_WORLD, 1) - process 29 OR I have also seen errors the same as above but arising from “atom:improper_atom” or “atom:dihedral_atom”. I tried to make working example with a simplified version of my simulation but I was not able to reproduce the error for smaller systems. I have no clue what triggers such a behavior: is it LAMMPS issue? or something with the cluster that I’m running on? or my input file parameter settings? I’m attaching the input file that I use for further clarification of what I’m doing, however, the data file for the system is huge (almost 70 MB) but I can provide it if it’s needed for further investigations. Best, Kasra.
delete_bonds-issue.in (2.92 KB)
