I have taken these parameters from “Effect of Adsorbed Alcohol Layers on the Behavior of Water Molecules Confined in a Graphene Nanoslit: A Molecular Dynamics
Study”. I have seen a lot of papers using the oplsaa force field. Can you suggest what is wrong with simulation parameters.
I have taken these parameters from “Effect of Adsorbed Alcohol Layers on the Behavior of Water Molecules Confined in a Graphene Nanoslit: A Molecular Dynamics
Study”. I have seen a lot of papers using the oplsaa force field. Can you suggest what is wrong with simulation parameters.
i am not a magician nor a mind reader, so i cannot give you a specific answer to this. also, i am not questioning the publication. but how can you be certain, that you made no mistake in entering them or other simulation parameters? MD simulations follow the GI-GO principle (i.e. garbage in, garbage out), thus if you get garbage results, chances are very high, that there was garbage already in the input, and there are many ways to mess up MD simulation inputs. however, it is impossible to say from so limited information where this happens. experience has shown, that people do make mistakes, especially people that present their questions in the way you are doing it.
axel.
Have you checked whether butanol can be found in the database of LigParGen ?
http://zarbi.chem.yale.edu/ligpargen/
Try to use the parameters from there, or compare to the one that you have in the ZZZ.in.settings.
That might give you a clue where the error is.
It is hard to say where the error with the piece of information that you gave.
Finally, I do not possess a large experience with lammps neither opls,
so it is up to you to figure out what you should do next.
I think the error in density is because the system is not neutral that can be seen in the attached screen shot. But if we see the topology file then each molecule is neutral and also i have checked the system.data file of butanol and found every molecule is neutral. I want to know how can i make the system neutral.
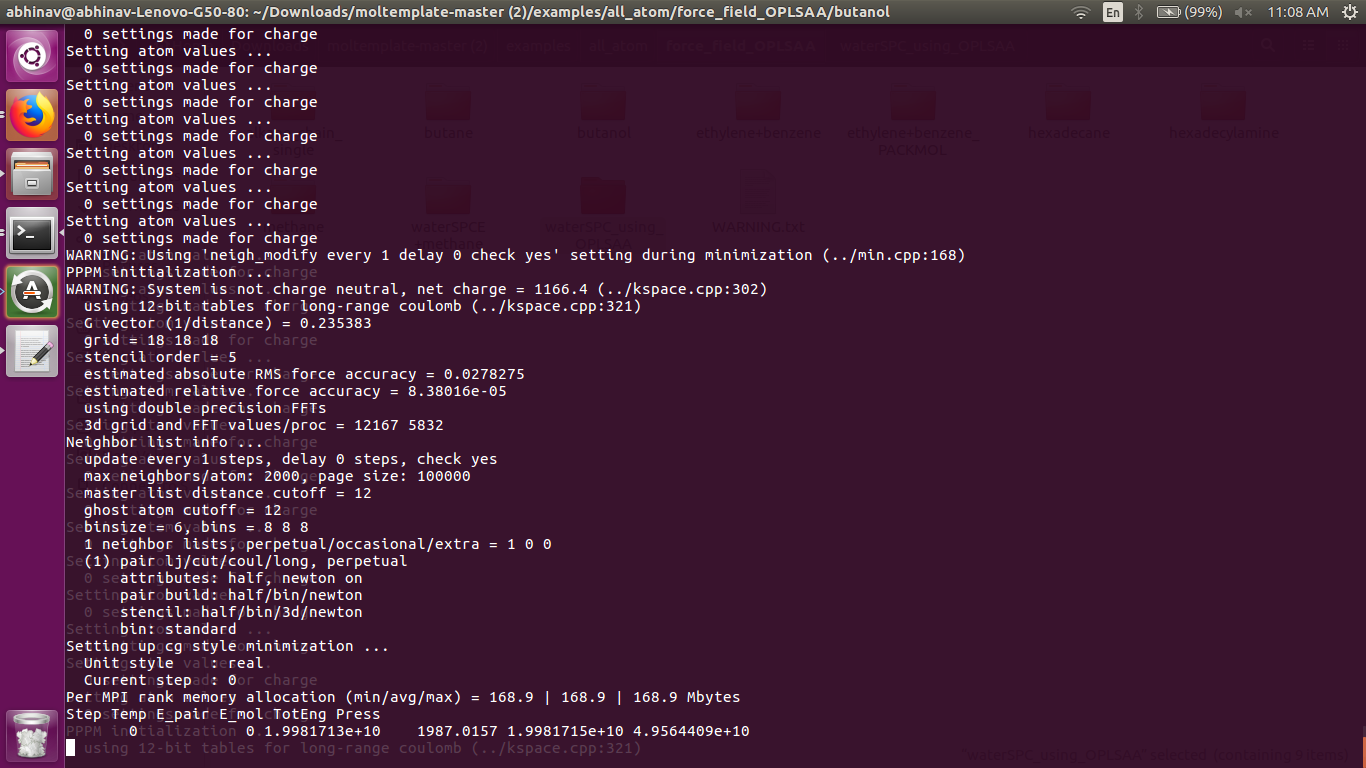
I think the error in density is because the system is not neutral that can be seen in the attached screen shot. But if we see the topology file then each molecule is neutral and also i have checked the system.data file of butanol and found every molecule is neutral. I want to know how can i make the system neutral.
like i mentioned before, this just confirms my GI-GO suspicion. the system is not neutral because something you do in your data file or input of force field settings is bogus. you have a gazillion of “0 settings made for charge” outputs. that can’t be right either.
in short, you don’t need to neutralize your system, you have to set it up correctly, which you obviously don’t. and since you are still not providing any helpful information for that, but keep claiming, that everything you do is correct, nobody can help you.
axel.