Dears
I want to measure density profile for a system water/pdms using MD with NVT and in fact I got it but I cant not get symmetric curve I mean the curve not start at zero and end to zero as I seen in most published paper. The input script and data are attached, am looking for some noticed that may help me
your force field is (still) bogus (I have commented on that multiple times and won’t repeat it)
your simulation settings are bogus (ditto)
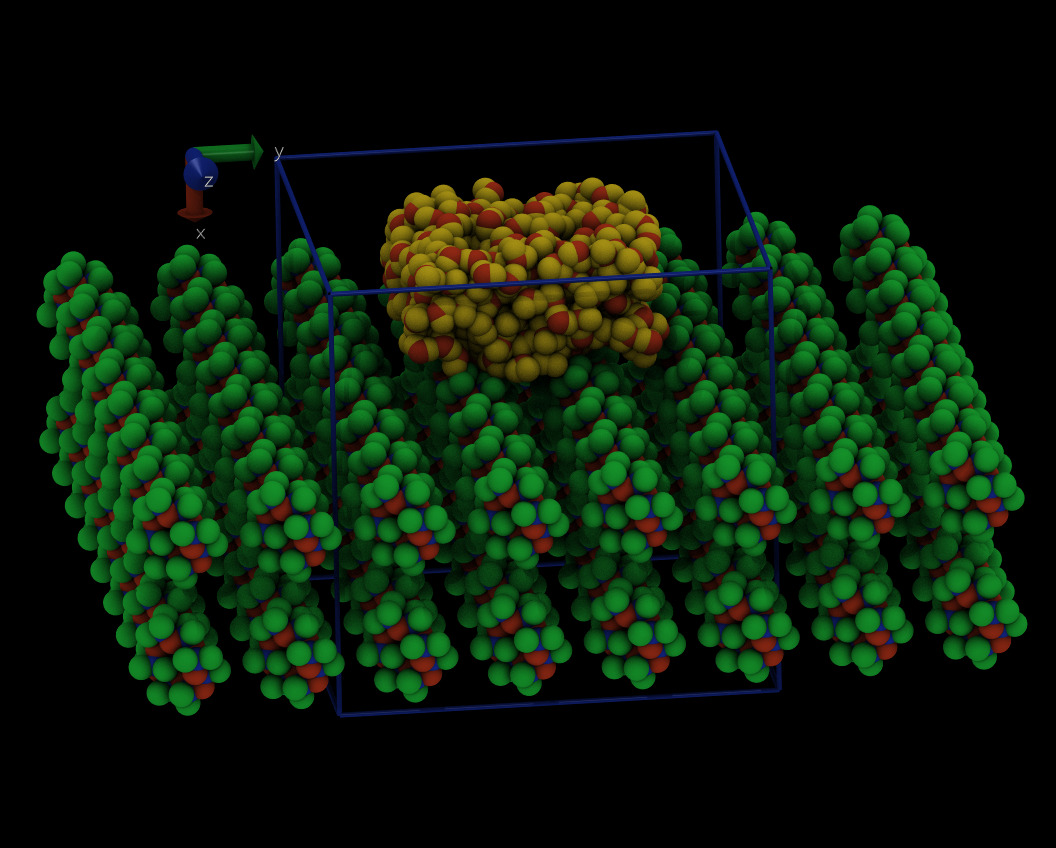
you are trying to plot the density profile in z-direction, but your layers are stacked in x direction (see image attached below).
Below is a visualization of your system as defined in the data file. A large part of it is outside the central simulation cell and will thus be wrapped back into the principal cell by LAMMPS when applying periodic boundaries. Please also note the axis labels.
Below is a visualization with one level of periodic images added in x- and y-direction colored in gray. You can clearly see how this creates a very bogus geometry, which is the main reason why you have unphysically high energies and pressures in your system.
Not getting the density plot you expect should be the least of your worries. What does it matter, if your density plot does not look like you expect when you are plotting bogus data. Since the last time I commented on a message of yours, you have made zero progress on the fundamental problems of your simulation thus highlighting - again - how bad an idea it is to keep doing this without proper tutoring and training and without following the advice being given.
Many thanks for your advices and help, according to your notes I followed your advices and I change force field, Geometry, setting, etc as in attached file. For new script I got Fluctuation in pressure from plus to minus please can you advise me , really I appreciate It. Am sorry in.input.9.lmp (6.7 KB) pdms-h2o.data (661.3 KB)
I got slow learning into lammps due to very few experiences in my collogues in Iraq. Hope you understand me.
Regards
No, I don’t understand, and it seems that you don’t fully realize how little understanding you have of what you are doing. That is, understanding of force fields, understanding of statistical mechanics, understanding of how to build and test molecular geometries, understanding of how to test and debug MD simulations. You are lacking crucial skills almost everywhere and - which is the most worrisome observations - you don’t seem to be able to improve on your own from the guidance you have been given in the last months.
Your input is still bogus and by a large margin. You just keep making some changes, but they appear rather random instead of motivated by science and are not directed at addressing the fundamental problems. There are far too many settings and parameters and choices that there is any hope you may hit the correct ones just by chance.
If you cannot get proper training, why do you keep doing things where you are destined to make gross mistakes? You are just setting yourself up for continuous frustration and public humiliation.
Just look at the crazy high potential energy of your system. That cannot be right.
Just a random observation: each of your water molecules has a net charge of -0.76. That cannot be correct.
After so many failed attempts to get a meaningful simulation going you have to own up to reality and make a decision: either you find a properly skilled (and patient) tutor and get the necessary training or you find a different area of research where there are sufficient skills available.
I for my part have no interest to provide any more responses to questions on secondary issues when there are so many fundamental mistakes.