Using 23Jun2022 version of LAMMPS, I have implemented the fix deposit command but the behavior is a bit odd. There are two major concerns I have: sometimes atoms don’t seem to be inserted in my specified deposition region, and there is a lot of melting occurring and I am unsure how to mitigate it. If the melting is a result of the addition of atoms to the bulk instead of the surface, then ok.
in.deposit (1.5 KB)
Generally, to help you debug, it’s helpful for us to see the output and/it log file of the simulation, to verify the behavior and look for other various issues.
The first thing I do notice, though, is that your deposit region is pressed right up against your surface. As atoms get deposited, they’ll come to a rest inside this region, and therefore might interfere with the placement of new atoms. Normally there would be a separation between the surface and the deposit region.
To this end, I would increase the z-height of your box, separate the region by enough space to accommodate the additional atoms, and try again. If that doesn’t fix all your issues, then give us your updated input script and output.
1 Like
I tried moving the deposition region away from the surface like you suggested, but when I visualize with OVITO, I still get atoms being deposited in the bulk (towards the bottom of the block) for some reason. I also get a warning about particles being time-integrated multiple times, but that shouldn’t be the case? Is there something I’m missing about how groups work?
in.deposit (1.5 KB)
log.lammps (80.6 KB)
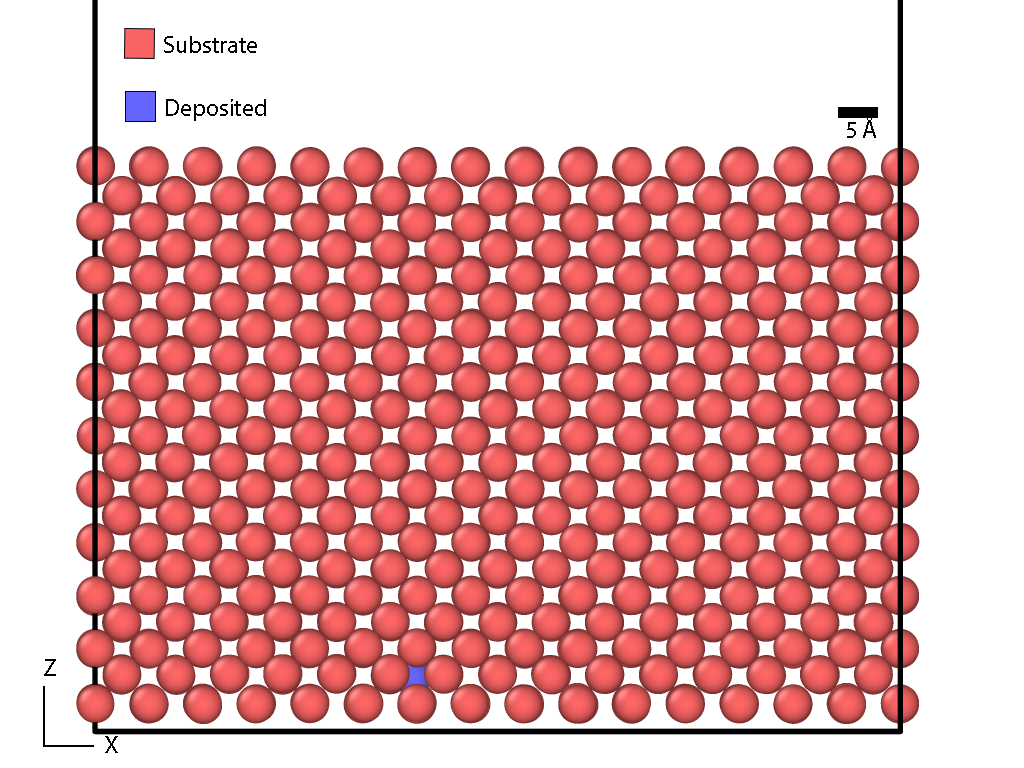
It looks like the groups may have some intersection, based on the number of atoms in each. 1125+2925+1125 (bulk+langevin+surface) = 5175, not 4175, so there’s overlap somewhere. Your lattice probably falls directly on z=2 or 8. That overlap would cause the integration error.
FWIW, the ambiguity of lattice positions on integer region boundaries can be easily avoided by either using non-zero non-integer values for the origin keyword with the lattice command or by using non-integer region boundaries.