Hello everyone,
I am currently working on a model where I am using tabulated bonded potentials, tabulated angle and non-bonded potentials. I am not sure how to implement the fixes for my simulation model. The condition is as follows:
Ensemble information:
first 5 nano seconds --> NVT
for the remaining 120 ns --> NPT
temperature information:
0 to 25 ns --> constant temp. of 590K is maintained
25 ns to 125 ns --> temperature decreases by 10K at every 2 ns
I have tried implementing NVT fix in my input script, but I am not sure where to put the time information in it. How should I proceed? Any leads would be appreciated.
I hope I was able to convey my problem clearly.
Thank you for your time.
Kind regards,
Vinay

You can run a series of short simulations with different thermostat
and barostat parameters by using the "unfix" command.
I attempted to provide examples of usage below.
I am not sure how to implement the fixes for my simulation model. The condition is as follows:
Ensemble information:
first 5 nano seconds --> NVT
units real
timestep 1
# 1timestep = 0.001ns (assuming "units real")
fix 1 all nvt temp 300.0 300.0 100.0
run 5000
unfix 1
for the remaining 120 ns --> NPT
temperature information:
0 to 25 ns --> constant temp. of 590K is maintained
25 ns to 125 ns --> temperature decreases by 10K at every 2 ns
timestep 1 # 1timestep = 0.001ns (assuming "units real")
fix 1 all npt temp 590.0 590.0 100.0 iso 100.0 100.0 1000.0
run 25000
unfix 1
fix 1 all npt temp 580.0 580.0 100.0 iso 100.0 100.0 1000.0
run 2000
unfix 1
fix 1 all npt temp 570.0 570.0 100.0 iso 100.0 100.0 1000.0
run 2000
unfix 1
fix 1 all npt temp 560.0 560.0 100.0 iso 100.0 100.0 1000.0
run 2000
unfix 1
:
etc...
Of course, feel free to alter the timestep (1fs) and pressure
(100.0bar) as well as the Nose-Hoover thermostat & barostat parameters
(100.0 and 1000.0) to fit your needs. You might want to rename "1"
(in "fix 1") to something else "myfix" or "fxMove".
Cheers
Andrew
My apologies. In my last email I wrote had some mistakes:
units real
timestep 1
# 1timestep = 0.001ns (assuming "units real")
somewhat embarrassing. That should have been 1timestep = 0.000001ns
(So I should have increased the lengths of all of the "run" commands
accordingly by x1000).
This is the kind of message which is not that helpful to anybody, but
I feel the need to post the correction anyway because of pride.
You can run a series of short simulations with different thermostat
and barostat parameters by using the "unfix" command.
LAMMPS Molecular Dynamics Simulator
I attempted to provide examples of usage below.
>
> I am not sure how to implement the fixes for my simulation model. The condition is as follows:
>
> Ensemble information:
> first 5 nano seconds --> NVT
units real
timestep 1
# 1timestep = 0.001ns (assuming "units real")
fix 1 all nvt temp 300.0 300.0 100.0
oops, I just realized you wanted a starting temperature of 590K
fix 1 all nvt temp 590.0 590.0 100.0
run 5000
run 5000000
unfix 1
> for the remaining 120 ns --> NPT
>
> temperature information:
> 0 to 25 ns --> constant temp. of 590K is maintained
> 25 ns to 125 ns --> temperature decreases by 10K at every 2 ns
timestep 1 # 1timestep = 0.001ns (assuming "units real")
fix 1 all npt temp 590.0 590.0 100.0 iso 100.0 100.0 1000.0
run 25000
run 25000000
unfix 1
fix 1 all npt temp 580.0 580.0 100.0 iso 100.0 100.0 1000.0
run 2000
run 2000000
Hello Andrew,
Thank you so much for such a detailed response. I get your point.
However, I have another issue related to the same model.
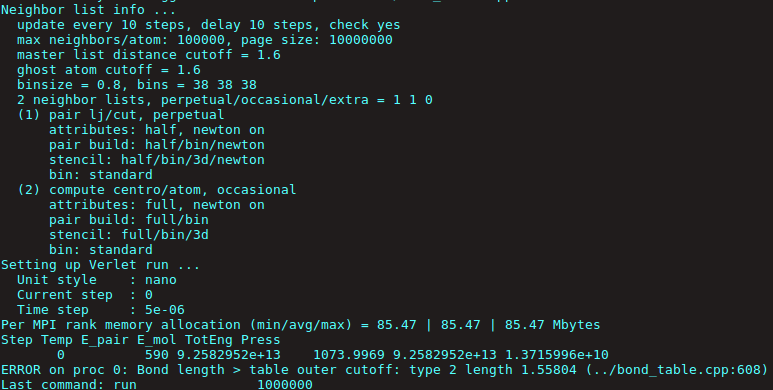
Since I am using tabulated bond potentials, my simulation throws an error at the very first time step that “Bond length > table outer cut off” type 2 length 1.55804.
The data I have in my tabulated potentials is only till 1.5 nm bond length. So as soon as the bond length exceeds this limit of 1.5nm, it throws an error.
I am attaching my tabulated data file, kindly have a look at it. (2nd column: bond length, 3rd: potential values, 4th: force as a negative derivative of potential)
Do you have any suggestions to debug this?
I am attaching a snapshot of what the error looks like.
Thanks once again. It really means a lot.
Kind regards,
Vinay

Coeff_Bond_Table_neg_d (391 KB)
To prevent the error, either reduce the temperature, or change the
shape of the energy&force function in the table file so that such
large distances between bonded atoms are not easily attainable.
You can lookup the meaning of error messages here:
https://lammps.sandia.gov/doc/Errors_messages.html
In this case,
"Pair distance > table outer cutoff
Two atoms are further apart than the pairwise table allows."
Hi Andrew,
The problem is I can change neither the temperature nor the energy & force function. I was wondering if there’s a way to check which bond (or two particles) is exceeding the limit of 1.5 nm. What do you think?
Regards,
Vinay

Even if you cannot change the shape of the energy curve, you can always extend the length of the table to handle larger radii. You can manually add one more line to the table file at a radius which is larger than you think is possible, and make sure that the force at this radius is a relatively large negative number (so it points inward). This might not fix the problem if you have bad dynamics. In that case you should visualize your system.
More response below…
The problem is I can change neither the temperature nor the energy & force function. I was wondering if there’s a way to check which bond (or two particles) is exceeding the limit of 1.5 nm. What do you think?
some relevant links to this question:
https://www.google.com/search?q=lammps+bond+table+outer+cutoff
https://lammps.sandia.gov/threads/msg78797.html
I also strongly suggest looking at your simulation results visually.
One way to do that is to use VMD. I attached some instructions for doing that to this message (“README_visualize.txt”).
Try to save the trajectory often enough that you can hopefully see when the bond is getting stretched.
Cheers
Andrew
README_visualize.txt (2.86 KB)