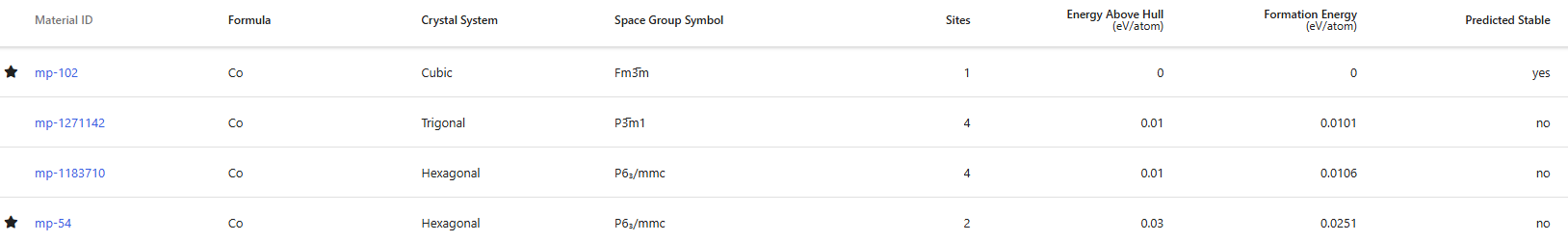Thank you so much for your response.
And I’m sorry for checking late and replying now.
Somehow, I managed to successfully extract the data.
I’m sorry for keeps asking, but I still don’t really get it and there are parts that keep confusing me…
- You don’t need to call
mpr.get_structure_by_material_id(). The structure should already be in your structures list and accessible via structure_summary.structure
- Does this mean that since
structure data is already inside structures, we don’t need to call structure = mpr.get_structure_by_material_id(material_id) again?
- So, does that mean I can just use
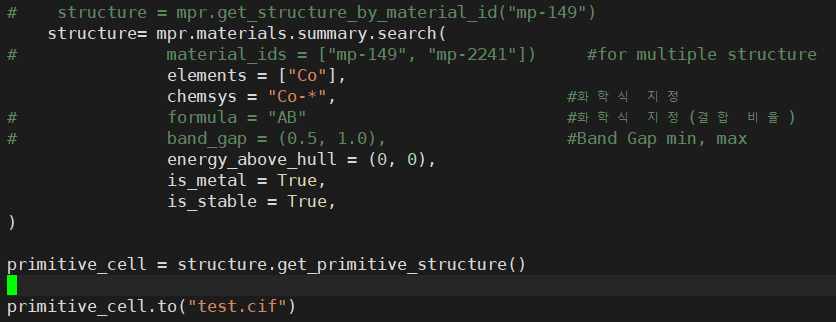
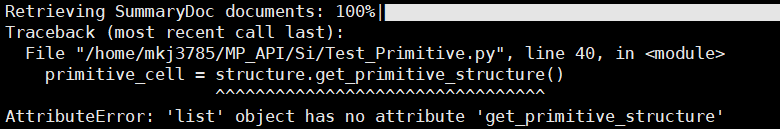
structures = mpr.materials.summary.search(~) to extract the data without necessarily using primitive_cell = structure.get_primitive_structure(), and it will still give me the primitive cell structure?
- I’m asking if just using the second piece of code I posted above can extract the primitive cell.
Here I have a few more questions. Regarding the indicators provided by the materials project, are the sorting results for predicted stable and Energy above Hull the same?
I looked into the cases of Cu, Ru, and Co, and for metallic compounds (alloys), it seems that if Energy above Hull = 0, they are all considered predicted stable.
I know that generally, a state with Energy above Hull = 0 is thought as the most stable state. However, I’m wondering if there’s a specific reason for having those two options split into different columns.
Additionally, could you please direct me to detailed explanations for each column elements (synthesizable, is metal, predicted stable, formation energy, etc.)? I recall finding explanations for those criteria on the site before, but now I cannot locate them.
+ Is there any way to plot the cohesive energy of each materials?
I referred to dwinston’s answer and added the items below. (For MPRester, I’m just using the mp_api.client as it is.)
from pymatgen.ext.matproj import MPRester
mpr = MPRester()
mpr.get_cohesive_energy('mp-721988', per_atom=True) # 6.73 eV/atom
My python file is below.
import os
from mp_api.client import MPRester
with MPRester("ZiWsNasQOf0xOblqHoCg97K6Cu2kWk7B") as mpr:
structures = mpr.materials.summary.search(
elements = ["Co"],
chemsys = "Co-*",
energy_above_hull = (0, 0),
is_metal = True,
is_stable = True
)
with open("test.txt", "w") as file:
file.write(f"Number of documents: {len(structures)}\n\n")
if not structures:
file.write("No results found.\n")
else:
for idx, structure_summary in enumerate(structures):
material_id = structure_summary.material_id
formula = structure_summary.formula_pretty
sym = structure_summary.symmetry
structure = mpr.get_structure_by_material_id(material_id)
# cohesive = mpr.get_cohesive_energy_by_material_id(material_id, per_atom = True)
cohesive = mpr.get_cohesive_energy(material_id, per_atom = True)
primitive_cell = structure.get_primitive_structure()
cif_filename = f"{formula}.cif"
primitive_cell.to(filename = cif_filename)
file.write(f"--- Document {idx + 1} ---\n")
file.write(f"Material ID: {material_id}\n")
file.write(f"Symmetry: {sym}\n")
file.write(f"Cohesive: {cohesive}\n")
file.write(f"Primitive Structure:\n{primitive_cell}\n")
file.write('-' * 40 + "\n")
file.write("\n\n")
However, the following error occurs. Is it not possible to use mpr.get_cohesive in our mp_api.client as referenced above?
Traceback (most recent call last):
File “/home/-/MP_API/API/Test_Cohesive.py”, line 35, in
cohesive = mpr.get_cohesive_energy(material_id, per_atom = True)
^^^^^^^^^^^^^^^^^^^^^^^
File “/home/-/anaconda3/lib/python3.11/site-packages/mp_api/client/mprester.py”, line 399, in getattr
raise AttributeError(
AttributeError: ‘MPRester’ object has no attribute ‘get_cohesive_energy’
Best regards.