Hello,
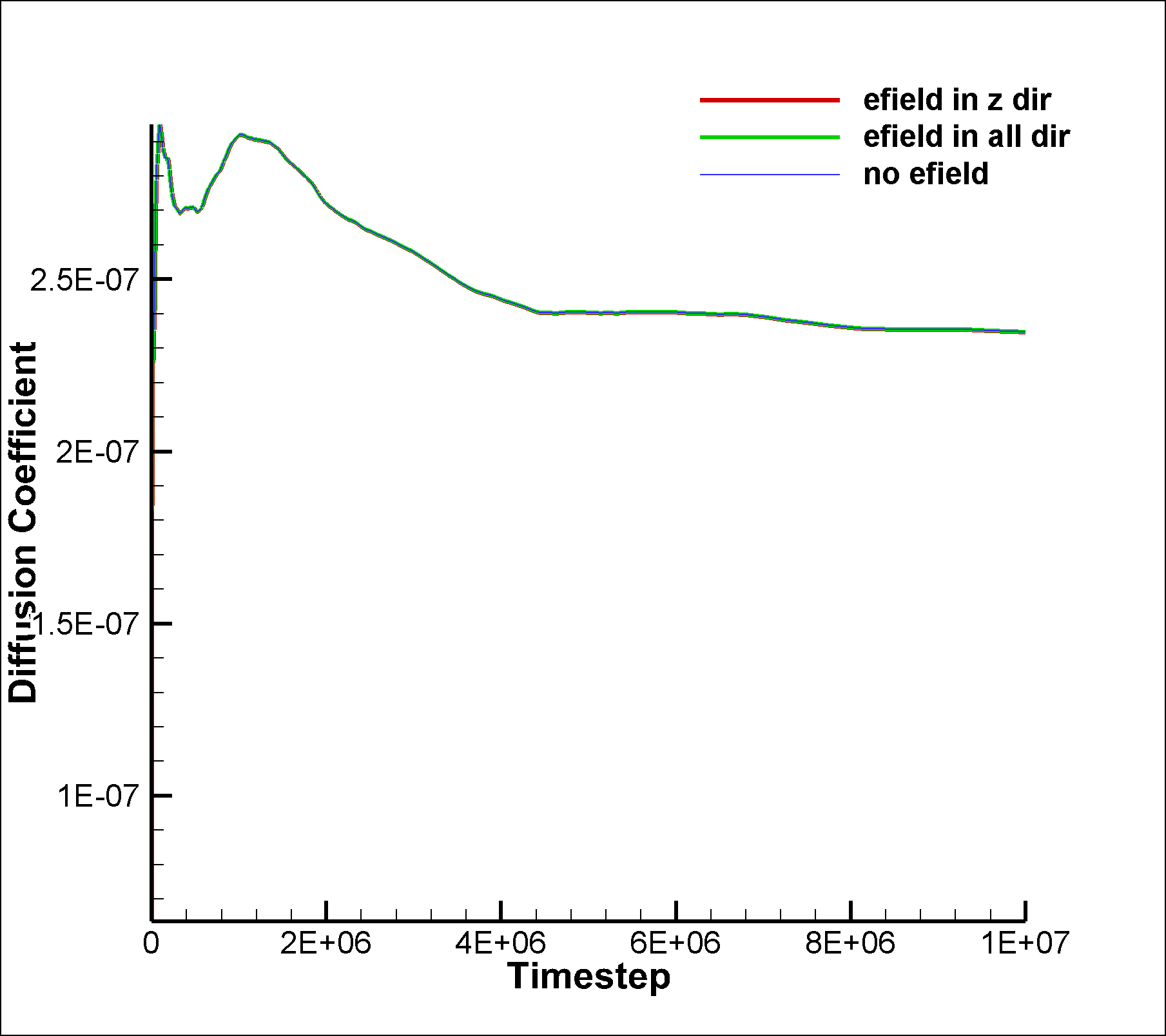
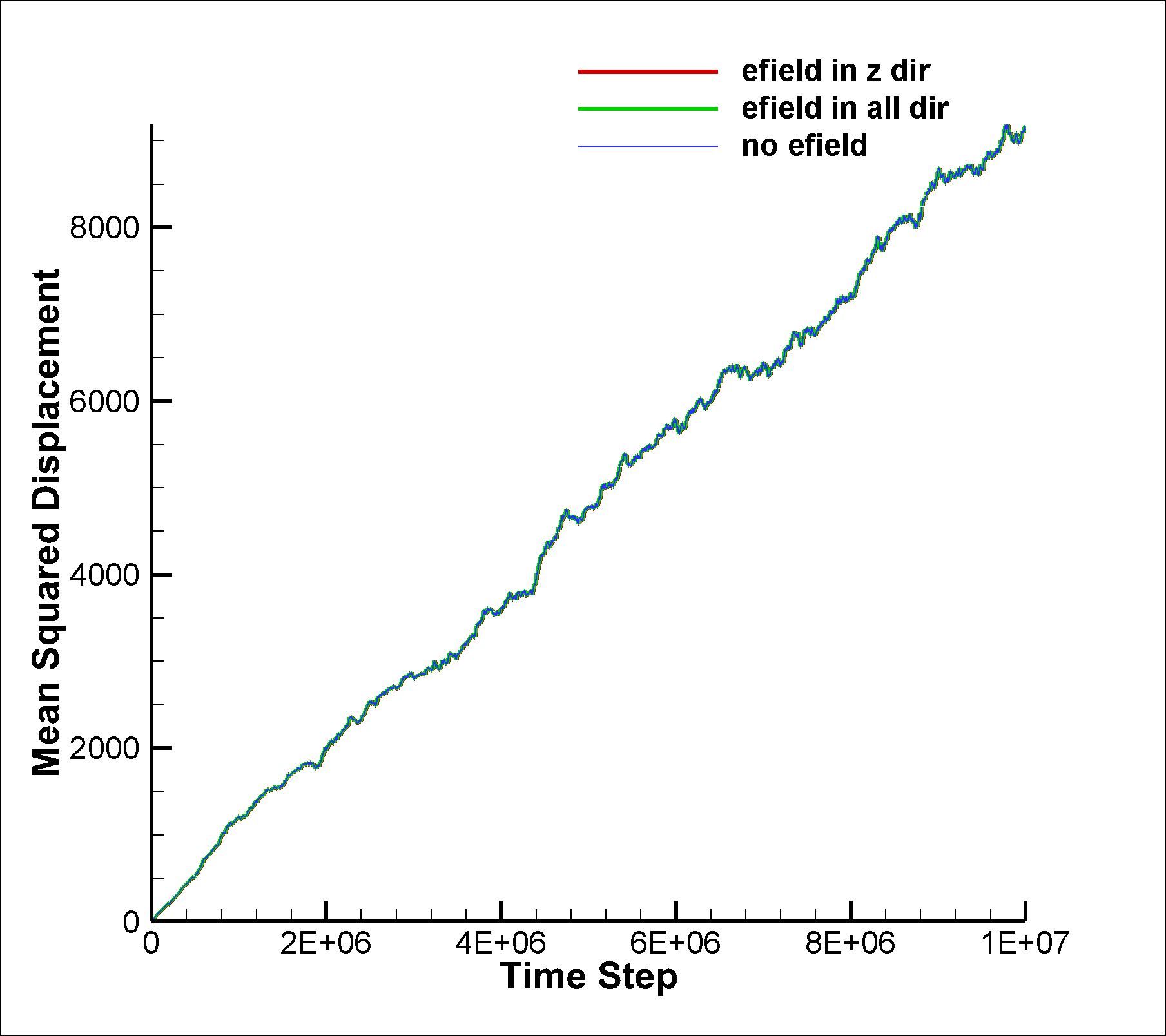
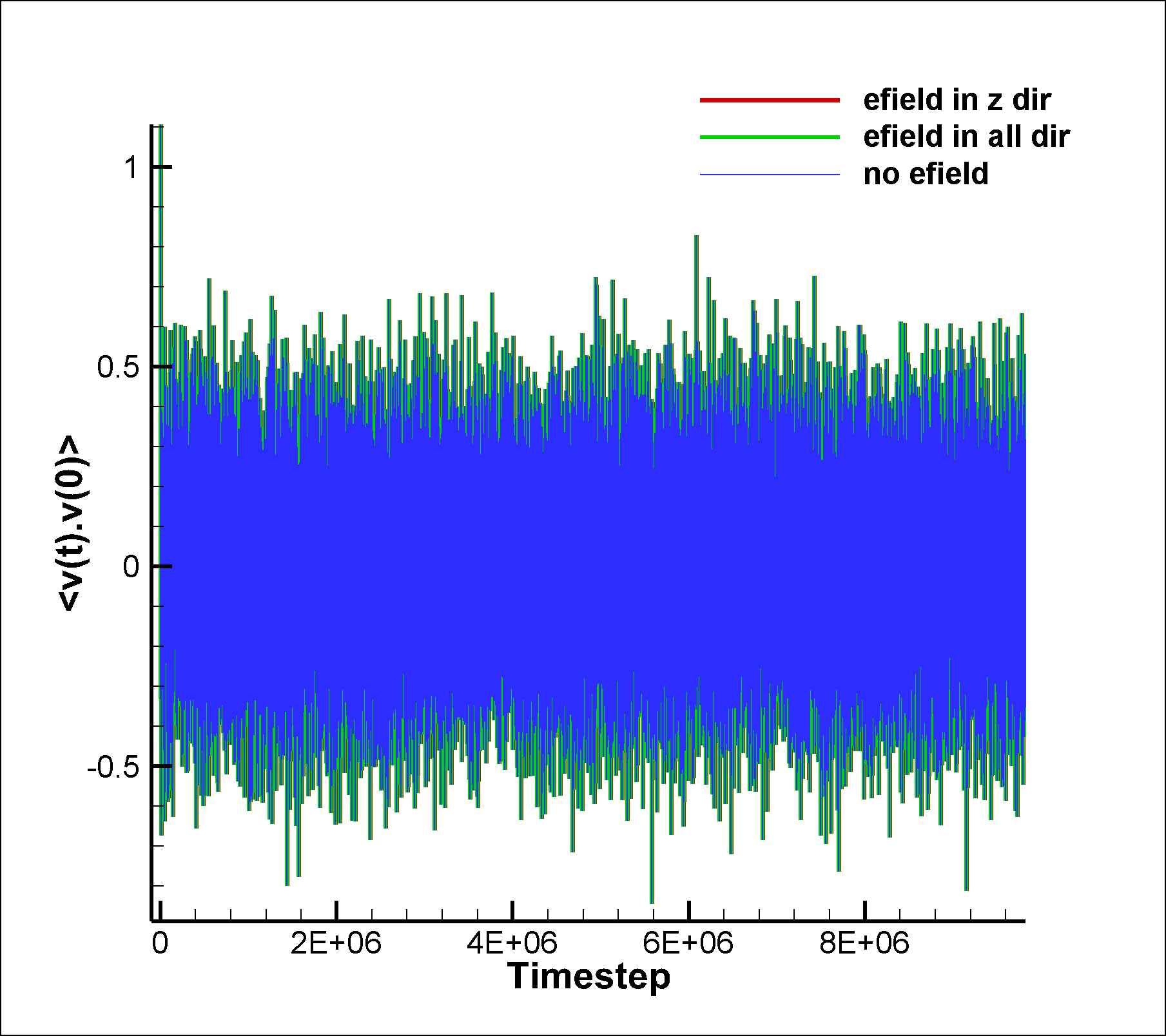
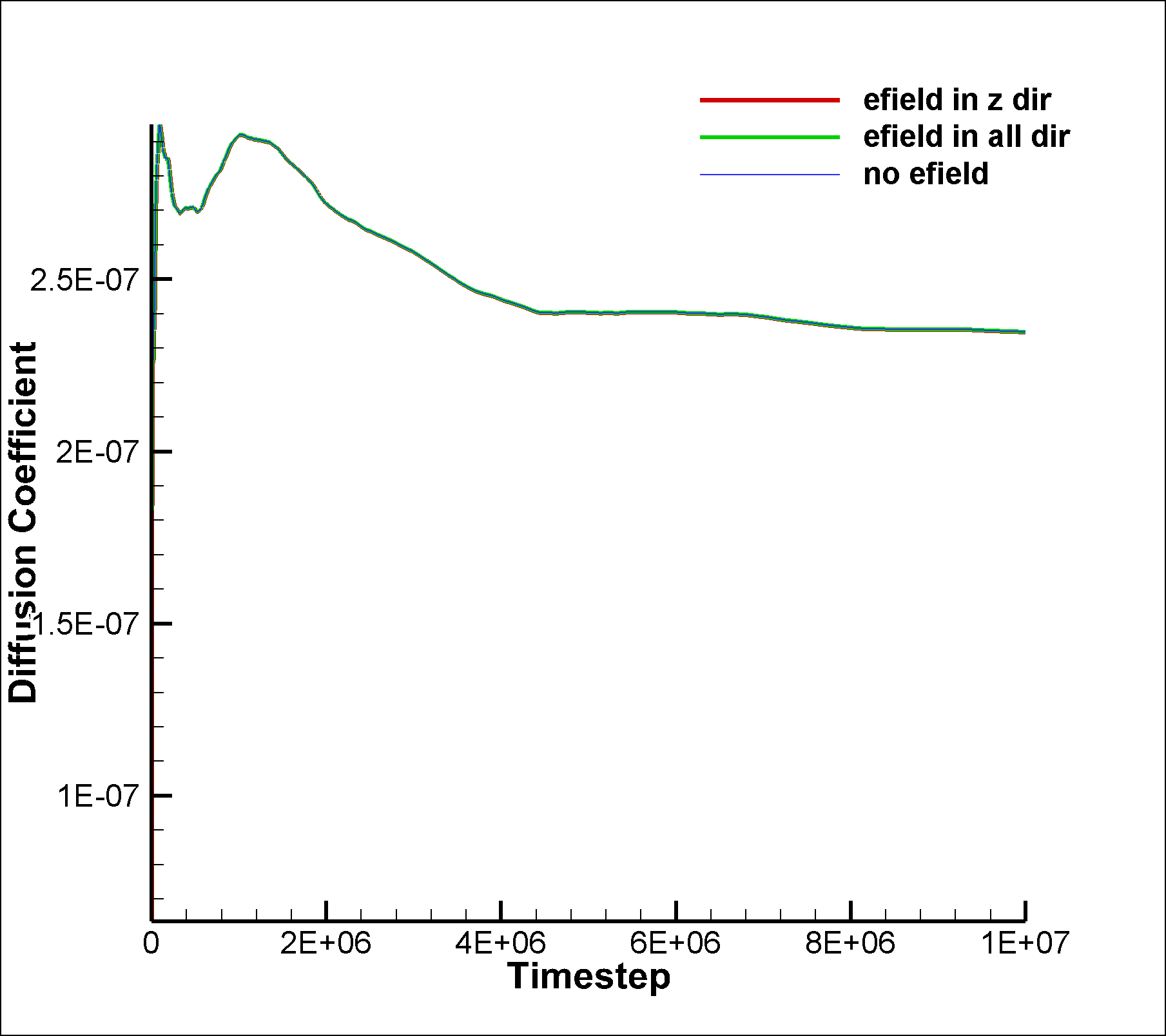
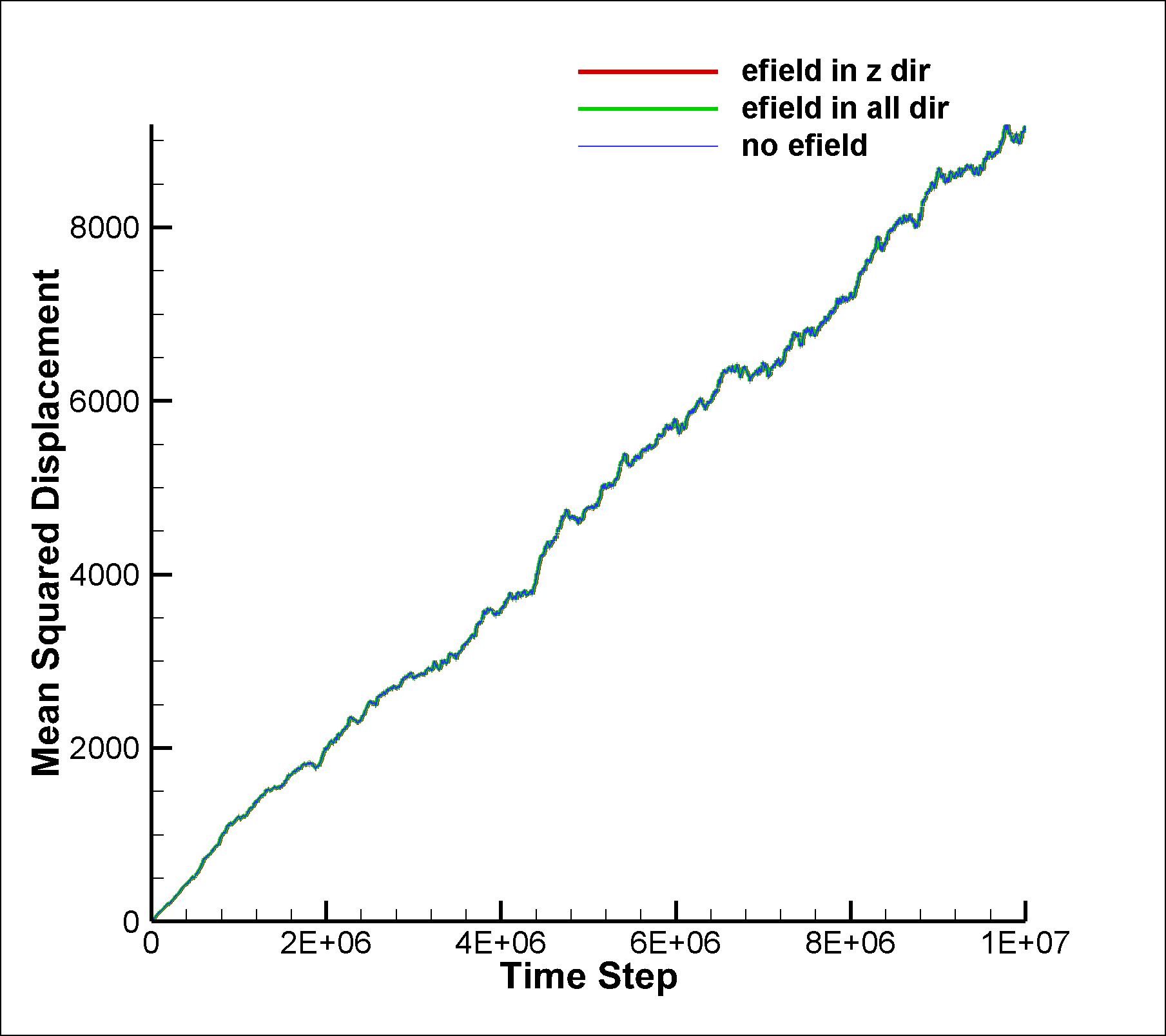
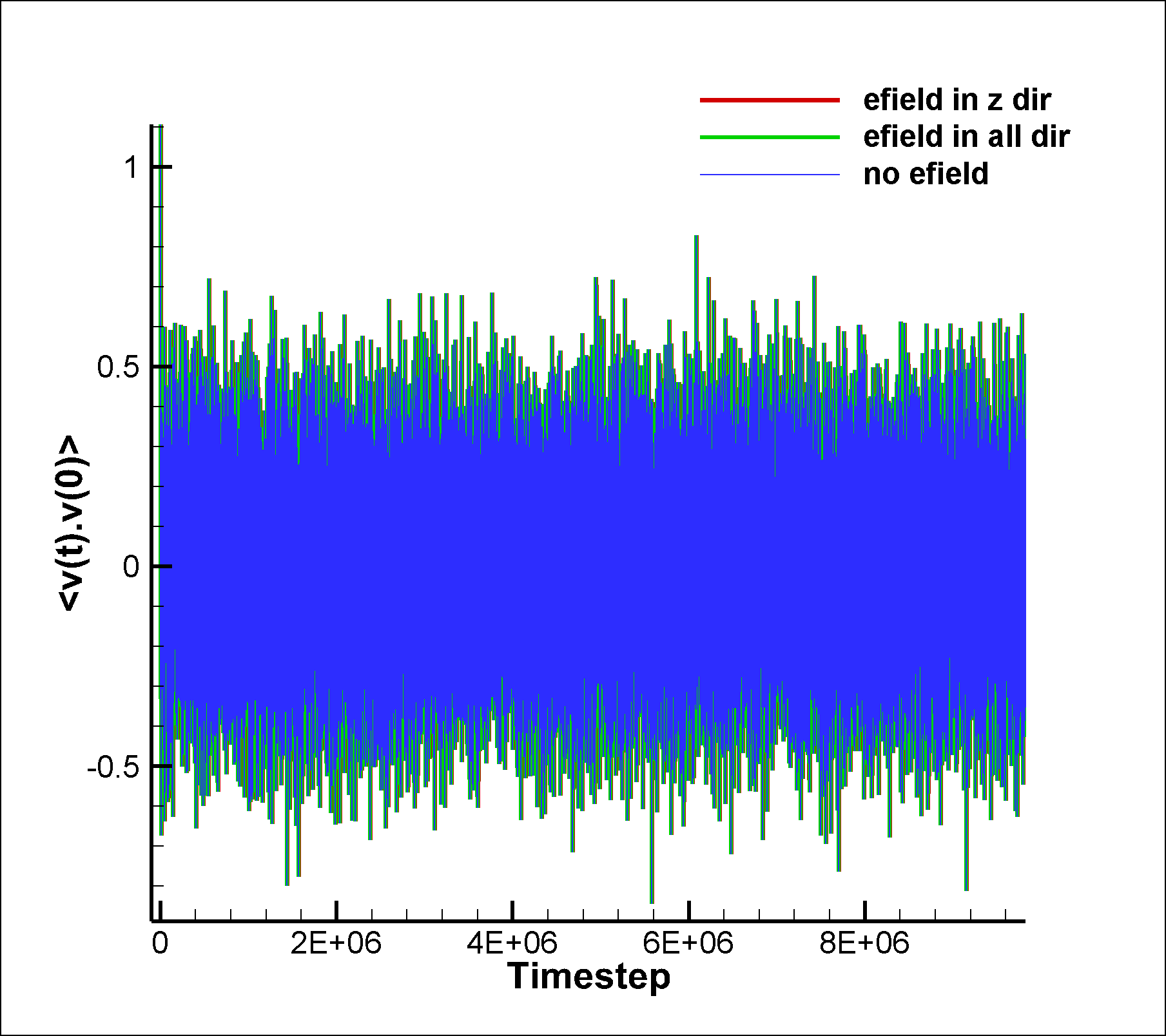
I am attaching two input scripts, one with a mixture SRD of style charge (initial charge set to 0.0) and the other with a net dipole of 1.0 (in reduced lj units). I expected to see an effect due to electric field on the system in the case of dipole, but I am not able to observe any difference between the results of the two runs. I am also attaching the graphs for vacf, msd and diffusion coeffcient calculated using msd.
I have used fix efield, but I am not sure if it is being called?
Any advice please?
Sincerely,
John
in.srd.mixture.charge_efield (3.2 KB)
in.srd.mixture.dipole_efield (3.21 KB)
Hello,
I am attaching two input scripts, one with a mixture SRD of style charge
(initial charge set to 0.0) and the other with a net dipole of 1.0 (in
reduced lj units). I expected to see an effect due to electric field on the
system in the case of dipole, but I am not able to observe any difference
between the results of the two runs. I am also attaching the graphs for
vacf, msd and diffusion coeffcient calculated using msd.
I have used fix efield, but I am not sure if it is being called?
as the fix efield documentation states, fix efield adds a torque to
particles with a (point) dipole.
however, there is nothing in your input, that is in any way reacting
to this torque (or to the dipole outside of fix efield).
axel.
I looked into the documentation and updated the pair_style to lj/cut/dipole/long to account for dipole-dipole interactions. I also changed from ‘fix nve’ to ‘fix nve/sphere update dipole’. The documentaion said that I need to include
kspace_style ewald/disp 1e-4
But I am getting an error that that command cannot be used in a 2D simulation. How do I effectively make this into a 3D simulation and get the same results? Or can is there a workaround to getting the above command working in 2d?
Thanks,
John
Attaching the input file.
in.srd.mixture.dipole_efield (3.37 KB)
I looked into the documentation and updated the pair_style to
lj/cut/dipole/long to account for dipole-dipole interactions. I also changed
from 'fix nve' to 'fix nve/sphere update dipole'. The documentaion said that
I need to include
kspace_style ewald/disp 1e-4
But I am getting an error that that command cannot be used in a 2D
simulation. How do I effectively make this into a 3D simulation and get the
same results? Or can is there a workaround to getting the above command
working in 2d?
please see kspace_modify slab.
axel.
In order to do this, I had to change the dimension to 3d and changed the lattice from sq to sc and boundary p p f. Since I cannot enforce2d in a 3d simulation, how do I zero out forces and velocities in z direction?
In order to do this, I had to change the dimension to 3d and changed the
lattice from sq to sc and boundary p p f. Since I cannot enforce2d in a 3d
simulation, how do I zero out forces and velocities in z direction?
velocity command and then fix setforce?
axel.
Thank you, it works now! I am getting a warning saying that kpsace is being invoked when there are no charges present. This is true since my input consists only of dipoles. Is it still necesary for me to use kspace_style?
Thanks,
John
Thank you, it works now! I am getting a warning saying that kpsace is being
invoked when there are no charges present. This is true since my input
consists only of dipoles. Is it still necesary for me to use kspace_style?
if you use a "/long" style, yes. for dipole-dipole interactions only,
it might be acceptable to use lj/cut/dipole/cut or lj/sf/dipole/sf
instead and forego the long-range electrostatic handling. but that is
up to you to decide what is handling the interactions of your system
sufficiently accurate.
axel.
My simulation keeps stopping because atoms are getting lost during minimization. I tried increasing the size of the domain, yet this is happening. I am attaching the log and input file.
in.srd.mixture.dipole_efield (3.38 KB)
log_efield.lammps (2.35 KB)
I also need to account for the long range interactions, therefore I am wondering if the box size in z direction being very large affects the simulation in any way.
there are far too many variables in your current setup. i would
recommend you take a step back and start with a small system, that has
only LJ particles (no SRD) and then you start with LJ-only, then LJ
plus cutoff dipole and finally LJ plus long-range dipole and adding
SRD only at the end. then you can see where things start to go south
and you can try to figure out why. as soon as you have the why, it
should be straightforward to decide what to do next. learning how to
do something that you haven't done before with a quite complex input
and system, is asking yourself for a lot.
axel.
I would say that using PPPM slab to model a 2d system
is problematic. That is really designed to model a 3d system
that is nonperiodic in one dimension (z), which is not
the same as an (infinitely thin) 2d system. I’m not sure
you can trick LAMMPS into thinking it is the same.
What is really needed is a 2d PPPM (or Ewald) solver,
which LAMMPS does not currently support.
Perhaps Stan (CCd) can comment on what this
would take.
Steve