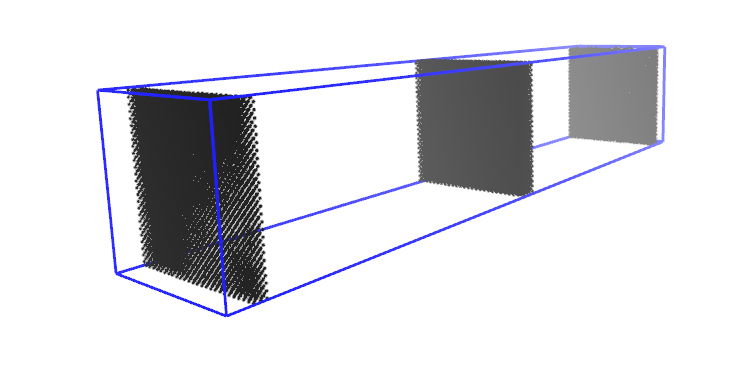
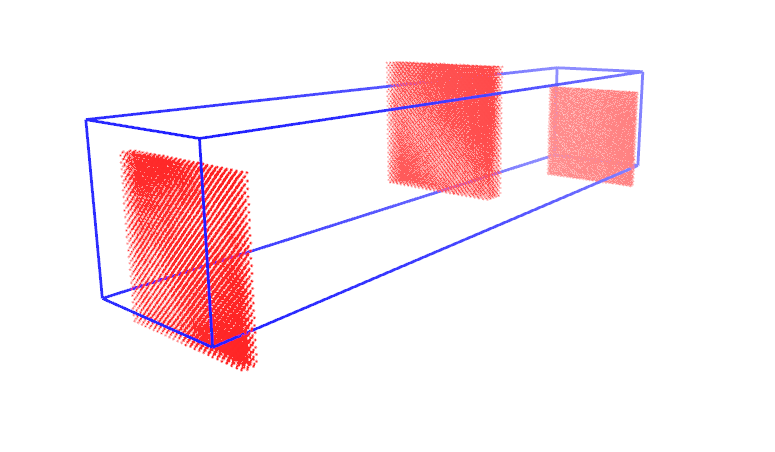
I am using lammps-16March2018, I built a simulation box including Pt atoms which are packed with FCC structure. As you can see in my input file, initially to equilibrate my system I am using Berendsen thermostat. To monitor my system evaluation, I write atoms coordinates in a dump file, besides, at the end of the simulation, I write the final coordinates of atoms in a data file. The problem is when I compare the last frame of dump file with the written data file (as you can see in the attached PNG files), in the case of data file some atoms crossed the simulation box, but in the case of the dump file, all the atoms remain inside the box. I also converted the written restart file to data file and it is similar to the data file written by write_data command.
I would be very thankful if someone could tell me why using dump command the final topology of the system looks different in comparison with write_data and restart commands!!
The thing is I want to start a new simulation using the previous equilibrated system and for me, it is important to know the exact atoms coordinate from the previous simulation.
####-------------------------------input file-----------------------------# ----------------------------------------------------------------------------- INITIALIZATION ----------------------
units real
dimension 3
boundary p p p
atom_style atomic
lattice fcc 3.91
#--------------------------------------------------ATOM DEFINITION-------------------------------------------------
processors 1 4 4
region PLt_left block 0 7.82 0 100 0 100 units box
region PLt_right block 490.7 500 0 100 0 100 units box
region PLt_middle block 240.18 259.82 0 100 0 100 units box
region whole block -15.0 515.0 0 100 0 100 units box
create_box 2 whole
mass 1 195.1
mass 2 39.95
create_atoms 1 region PLt_left
create_atoms 1 region PLt_right
create_atoms 1 region PLt_middle
group whole region whole
group PLt_left region PLt_left
group PLt_right region PLt_right
group PLt_middle region PLt_middle
timestep 1
#----------------------------------------------FORCE FIELD-----------------------------------------------------------
pair_style lj/cut 12
pair_coeff 1 1 7.49504 2.47
pair_coeff 1 2 0.157262 2.94
pair_coeff 2 2 0.238044 3.4
neighbor 2 bin
neigh_modify delay 0 every 1 check no
thermo 1000
thermo_style custom time temp ke pe lx ly lz press
thermo_modify lost warn
#####-----------------------------------------------------------Energy minimization
min_style cg
minimize 1e-16 1e-16 100000 100000
fix 1111 all box/relax iso 0.0 vmax 0.01
reset_timestep 0
timestep 1
restart 10000 restart.*.EQ
dump 1 all custom 1000 dumpsystem.lammpstrj id type x y z
velocity all create 300.0 4928459 rot yes dist gaussian
fix 2 PLt_left nve
fix 3 PLt_left temp/berendsen 300.0 300.0 100.0
fix 4 PLt_middle nve
fix 5 PLt_middle temp/berendsen 300.0 300.0 100.0
fix 6 PLt_right nve
fix 7 PLt_right temp/berendsen 300.0 300.0 100.0
run 500000
write_data data.Pt_Eq
Kind regards,
Shahin Mohammad Nejad