I want to calculate diffusion for water molecules in a system that has three layers. The layers are water - thick crystal - water, respectively.
I have found these two ways in the examples of Lammps. in.msd.2d.lmp (1.3 KB) in.vacf.2d.lmp (1.3 KB)
but the results that I obtain when I use them are not correct with both the methods.
My code is:
compute msd water msd com yes
variable twopoint equal c_msd[4]/4/(stepdt+1.0e-6)
fix 9 water vector 10 c_msd[4]
variable fitslope equal slope(f_9)/4/(10dt)
and also
compute cc3 water chunk/atom molecule
compute vacf water vacf
fix 4 water ave/time 1 1 1 c_vacf[4] #file tmp_vacf.dat
fix 5 water vector 1 c_vacf[4]
variable vacf equal dt*trap(f_5)
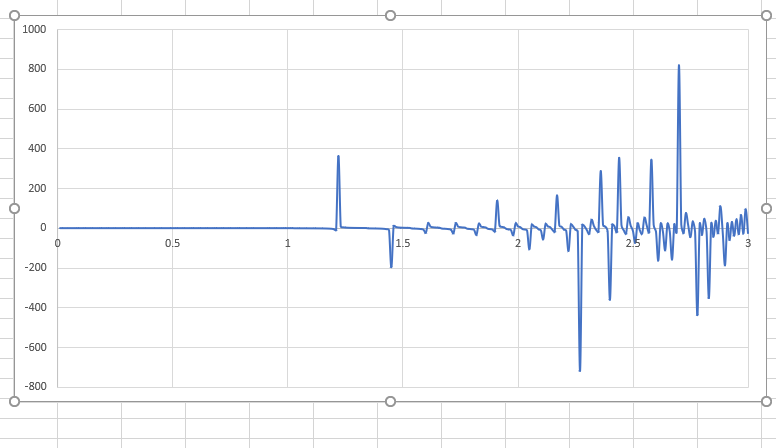
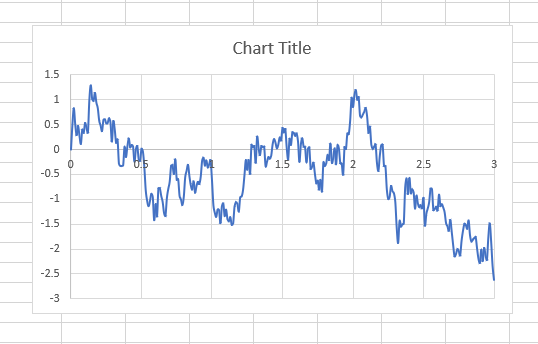
The results by msd method is figure named 1 and by vacf is figure 2. I have plotted the variables of two methods versus time (3 ns).
As I see in the examples of Lammps, the methods are for a pure system of water. I want to know in my system that the water layers are splited by a layer of crystal, are still the commands correct (both msd and vacf methods) or I should somehow change the commands so that Lammps computes diffusion for two water layers, separately? If yes, could you please tell what command I should add/change?
Thank you.
This is not so easy to answer with just some simple “do this, not that” kind of recommendations.
To begin with, if you read the README file in the LAMMPS examples folder, it will tell you that example inputs in folders with capital letters are “advanced” examples. That means, you need sufficient experience with creating and managing and analyzing simulations before attempting to use those examples and adapt them for your needs.
The example files you have are with “dimension 2” and “units lj” and for a simple Lennard-Jones 2d-fluid and not pure water. So those inputs will have to be adapted in many ways. To know how, you have to look up and understand how D is computed via MSD and/or VACF and what is different between 2d and 3d and also you need to adjust the units.
You typically would want to look at the diffusion of the center of mass, but for all practical purposes, you can just substitute the position of the oxygen atom for that.
Finally, computing self-diffusion for a non-homogeneous system is an even trickier business than for a bulk system, since the diffusivity is going to be different for atoms depending on the distance from the interface(s). So before even doing some serious computations, you first have to think about how to analyze something like this in general. Keep in mind that atoms/molecules are moving around.
Following the principle that one has to learn how to walk before trying to run, my suggestions are the following (which should be performed one step at a time):
adapt the example inputs from 2d systems to 3d systems. look up what results you should get instead and see how you have to adapt the computations. also see if you need to adjust the number of MD steps to get converged results.
check how you can improve the quality and accuracy of the results by averaging over multiple time origins
convert the input from reduced units to real or metal units and use parameters for Argon. Then look up what D should be in this case and try to reproduce it for the chosen conditions (try multiple)
convert from bulk liquid argon to bulk liquid water. pay special attention to how the size of the timestep and the number of timesteps need to be adjusted to have stable and accurate time integration with good energy conservation as well as getting converged results on D. The viscosity of water is different than that from Argon, so the settings are likely required to be different.
At this point you will have acquired the necessary skills to run analysis on the more complex “real” system, but you would still need to come up with a strategy to handle the case that diffusivity is position and direction dependent.