Dear Andrew, Paul and Everyone,
In the latest version of Lammps source code on the Dihedral Class2 part, there are several lines added by you in Jan 2013 to adjust the sign of phi.
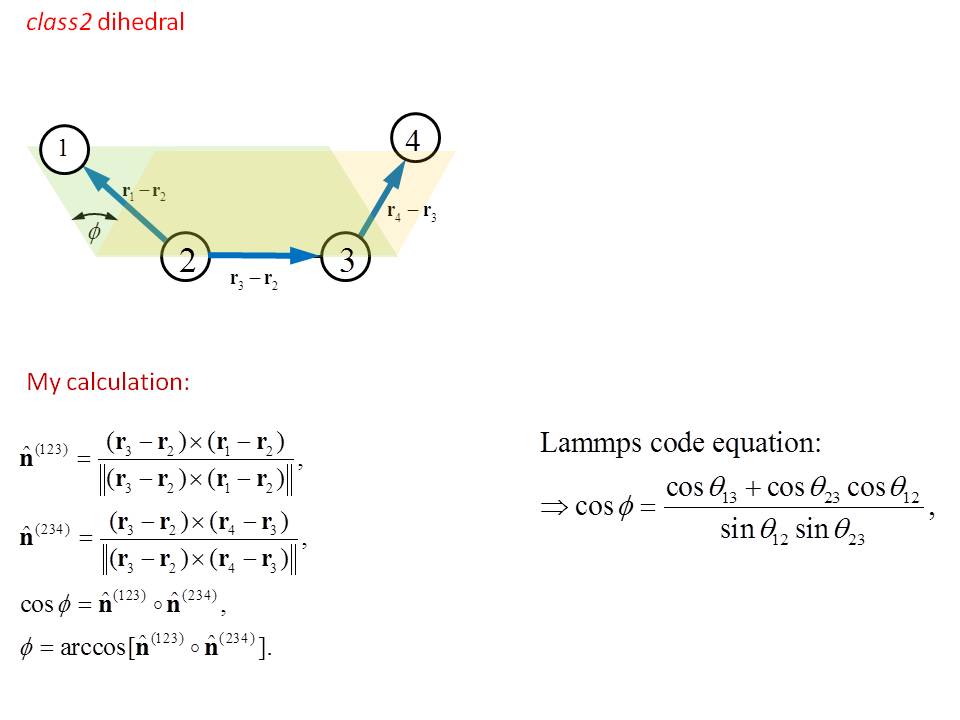
I have a question there: I calculated the dihedral angle using my own method as shown in the picture. It is the same as the equation in the code for cos(phi).
And it includes any geometry case of the four atoms.
I am not sure why that case should be considered separately and change the sign of the dihedral “phi”.
Is there anything more in the definition other than “the angle between plane 123 and plane 234 and the angle is in the interval [0 degree, 180 degree]?”
Thank you for your time,
Lili Zhang
Dear Lili
A distinction between positive and negative dihedral (and improper)
angles is necessary to design force fields for molecules that are
chiral. (Molecules which prefer one mirror-image over the other.)
Biopolymers, (see attached "1LMB3_ribbon.jpg") are made of homochiral
building-blocks (predominantly L-stereoisomers). In nature, both
stereoisomers (mirror images) may be equally stable (although the
barrier between the two may be high, preventing interconversion).
Consequently, it is probably not necessary to use these kinds of
forces for simulating realistic all-atom models of molecules.
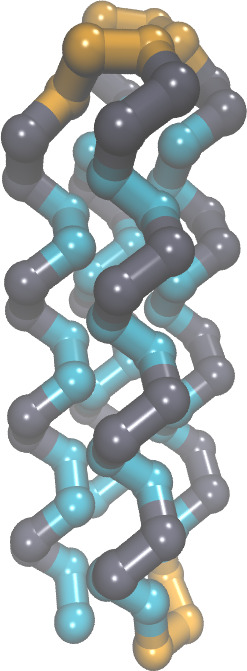
However much information is lost when using lower-resolution
coarse-grained models of molecules. (These models contain far fewer
atoms. For example, see the attached image "4HelixBundle.jpg". A
protein of that size would normally have about 1400 atoms.) Because
of the loss of detail, the force-fields are modified accordingly.
When this happens, the barrier separating stereoisomers may be too low
to prevent interconversion. Chiral force-fields are especially useful
for people who simulate coarse-grained biopolymers.
According to the documentation, class2 was at one point, the only
dihedral_style which supported both positive and negative dihedral
angles, and fractional phi0 (offset parameters). We had to fix this
because I knew people who were using class2 dihedrals to simulate
these coarse-grained protein models (and at high temperatures,
presumably seeing some crazy-looking left and right-handed helices).
Fortunately, now there's
http://lammps.sandia.gov/doc/dihedral_fourier.html
http://en.wikipedia.org/wiki/Chirality_(chemistry)
http://en.wikipedia.org/wiki/Homochirality
I hope this helps explain the motivation behind why I changed the code.
(It's true that in higher-than-3 dimensions, there is no way to make a
distinction between positive and negative dihedral angles. In
two-dimensions, you can define both positive and negative
bond-angles.)
Cheers
Andrew
Dear Andrew,
Thanks for your explanation.
We are using “realistic all-atom models of molecules” for polymers. Is it necessary to consider such adjustment for this case?
I also checked Lammps code for Improper Class2 calculation, and there is no such adjustment.
We are doing our own calculations for class2 force field and compare with Lammps results for the forces per atom. If it is really necessary to consider “positive” and “negative” dihedral (and improper) angles for our case, we will have to put those conditions in our calculation too.
Thank you for your help,
Lili Zhang
Dear Andrew,
Thanks for your explanation.
We are using "realistic all-atom models of molecules" for polymers. Is it
necessary to consider such adjustment for this case?
Hi Lili.
Just a quick email.
Technically speaking, you will need to worry about the sign of the
dihedral angle whenever you need to use non-zero "phi1", "phi2", or
"phi3" parameters (located in your dihedral_coeff commands, or in the
"Dihedral Coeffs" section of your data file).
However in practice, if K1, K2, and K3 are so large enough so that
the barrier between minima can not be crossed, then I suppose you can
ignore this issue.
I hesitate to bring up improper class2, because I am not very familiar
with it. I assume you will need to worry about this issue if you are
using non-zero X0 parameters.
(You might need to worry about it even if you use X0=0. I am not sure.)