Thanks for your information.
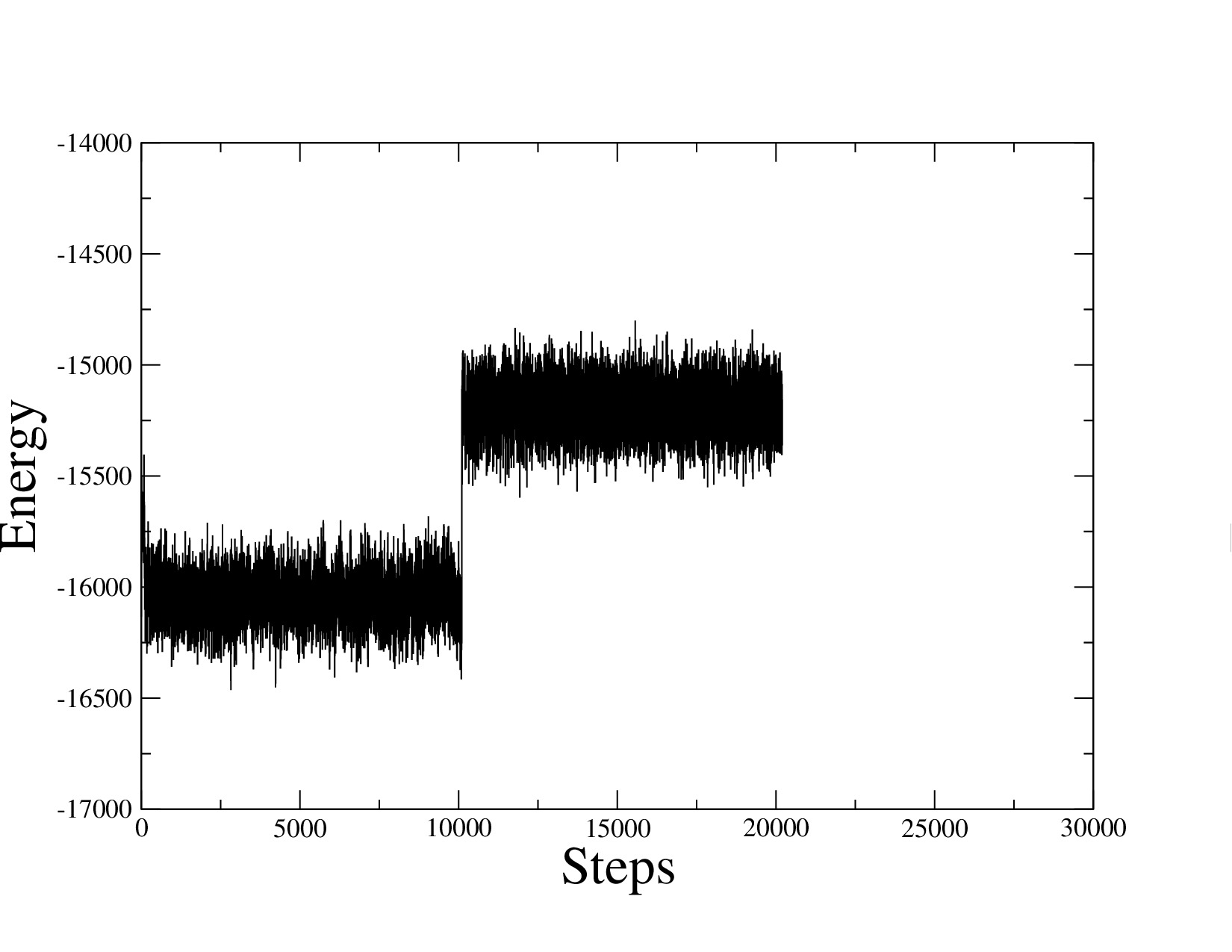
I tested the example file from lammps, and the energy of example system also increase when the time-step switch from 1fs to 2fs (show in figure).
I also built the system by EMC, and equilibrated the system via lammps. While I increased the time-step the energy of system increased either.
The example I chose is in.deca-ala_imd which located at /home/tntech.edu/czhan42/lammps-stable_11Aug2017/examples/USER/misc/imd
( I delete the fix shake commands and fix imd commond since I don’t need it)
When I searched on-line, I found something relate to energy drift.
However, I’m not sure if the lammps program show energy drift phenomenon. Is the properties result reliable with energy drift? Can anyone tell me how to eliminate the energy drift in lammps
Any comments would be appreciated.
Energy Fluctuations
[
Energy Fluctuations
](http://www.wag.caltech.edu/publications/theses/alan/subsection1_6_0_3_2.html)
Energy drift - Wikipedia
[
Energy drift - Wikipedia
](https://en.wikipedia.org/wiki/Energy_drift)
Kerwin
Tools like towhee, moltemplate, emc, AmberTools, (msi2lmp) use the atom type names of the participants atoms to generate angles, dihedrals, and impropers, as well as look up their force field parameters.
If some of of the dihedral parameters seems wrong to you, then it might mean that you have assigned the wrong atom type (type-name or type-integer) to some of your atoms. Unfortunately, depending on which tool you used to build your system, this can be easy to do.
In a force field like OPLSAA, there are dozens of different types of carbon, oxygen, and hydrogen atoms, for example. You must chose carefully.
(If you are using moltemplate, then you must read the description of each atom type in the “oplsaa.lt” file and chose the atom type carefully. For moltemplate OPLSAA examples, see https://github.com/jewettaij/moltemplate/tree/master/examples/all_atom/force_field_OPLSAA For details how to do this, see: http://moltemplate.org/force_field_recommendations.html )
If you are willing to try using other molecule builders, both “moltemplate” and “emc” support the OPLSAA force field. (Disclaimer: I wrote “moltemplate”)
If you know of a general tool that can read PDB files and automatically decide which type of carbon, hydrogen, oxygen… atoms should be used (from only the information in the PDB file), then please post a reply here.
Cheers
Andrew
P.S. It is possible to have OPLS dihedrals with ALL 4 of the parameters (K1, K2, K3, K4) set to zero. These dihedrals have no effect, but some force-fields insist on defining dihedrals every time 4 atoms are bonded together. These unnecessary dihedrals slow down the calculation slightly, but they are relatively rare. (They appear in less than 30% of the dihedrals in the OPLSAA force field parameter file. Eliminating them from the moltemplate force field file," oplsaa.lt", would solve this small problem, but it hasn’t been a priority for me to do that so far.)