I am using Moltemplate to generate a forcefield (L-OPLSAA) for my simulation. As a test run, I decided to make a forcefield for hexadecane from their given examples: moltemplate/examples/all_atom/force_field_OPLSAA/hexadecane.
After executing
moltemplate.sh system.lt
cleanup_moltemplate.sh
I get the following forcefield in the system.in.settings file:
pair_coeff 1 1 0.066 3.5
pair_coeff 2 2 0.066 3.5
pair_coeff 3 3 0.03 2.5
pair_coeff 4 4 0.026290630975 2.5
bond_coeff 1 268.0 1.529
bond_coeff 2 340.0 1.09
angle_coeff 1 58.35 112.7
angle_coeff 2 33.0 107.8
angle_coeff 3 37.5 110.7
dihedral_coeff 1 0.0 0.0 0.3 0.0
dihedral_coeff 2 0.0 0.0 0.3 0.0
dihedral_coeff 3 0.6446926386 -0.2143420172 0.1782194073 0.0
set type 1 charge -0.222
set type 2 charge -0.148
set type 3 charge 0.074
set type 4 charge 0.074
Dihedral type 3 is for atom types 1-2-2-2 which is (CT-C-C-C) where CT is the terminal carbon. Per the paper by Siu (https://pubs.acs.org/doi/10.1021/ct200908r), the dihedral parameters under for a fourier dihedral are (V0, V1, V2, V3) = (-0.0731, 0.64469263862, -0.21434201721, 0.1782194073) kcal/mol (see Table 2). These are decidedly NOT the parameters presented by moltemplate.
I wanted to ask if this is a bug in moltemplate? or is there a conversion i am not aware of over here?
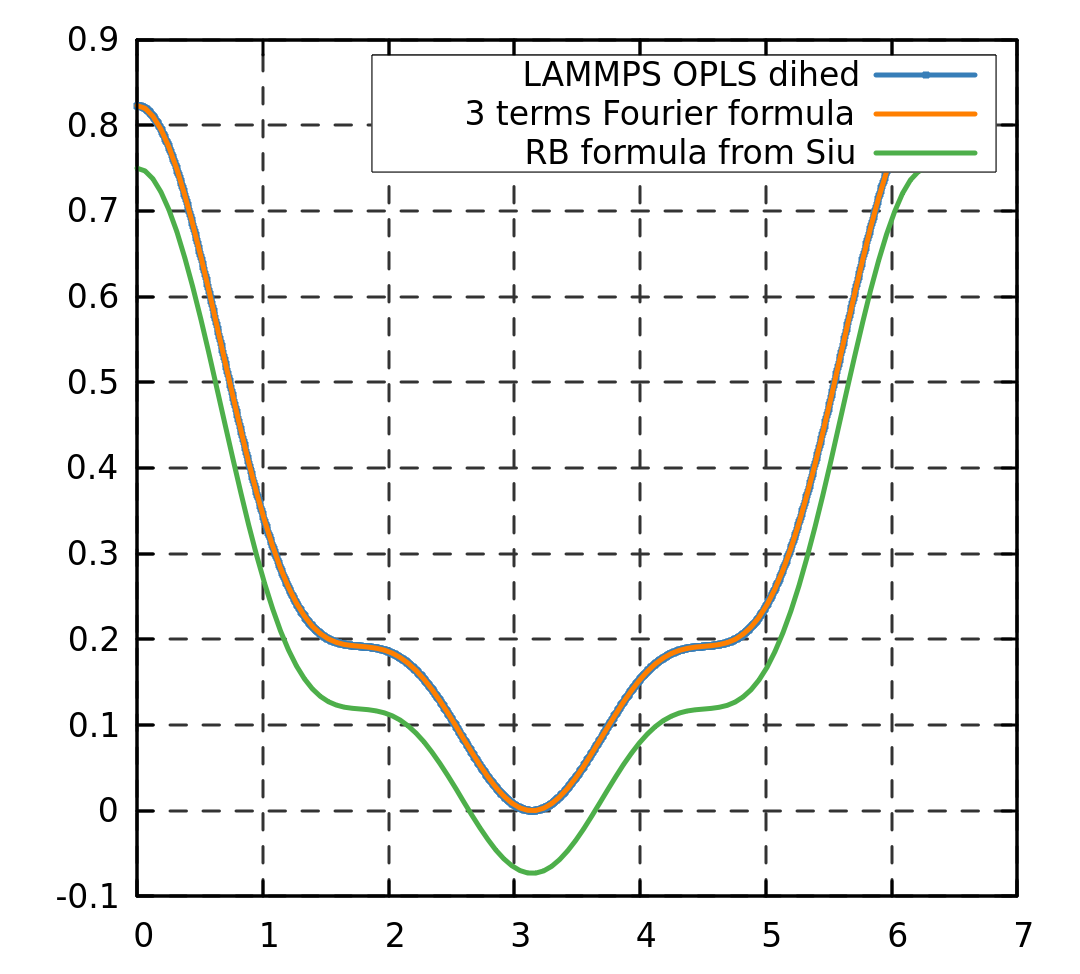
The dihedral conversion from one formula to another is generally a mess. The best way to know what is going on is to plot and compare. Unfortunately, the paper from Siu does not give the precise formula they refer to and the conversion, as they refer to a (probably outdated) version of the gromacs manual (see here for the current version).
As far as I can tell I do not know to what multiple of the dihedral angles correspond V_0, V_1 etc… However, plotting the RB formula is straight forward (if you don’t forget the difference of convention between \psi and \phi). I can reproduce the RB shape of the potential up to a constant using the moltemplate values and the 3 terms Fourier formula, so I think the parameters are correct. Note that the original OPLS formula used only 3 term for the dihedrals so it is not uncommon to have the last one of them set to 0. I suspect the V_0 parameter to be an energy constant that has no impact on the forces.
See an example of plot I got using the RB formula and your dihedral coefficient with the write_dihedral command.
Thank you for your response @Germain! This makes sense.
On the topic of moltemplate, though, @jewettaij, I wanted to ask why is there not C82L atom type? There is a C80L and a C81L atom type with the corresponding hydrogen types, but not one for C82L?
If you are interested in using the latest version of the force field, please consider writing a script for converting the parameters to the LT format and push your contribution to the Moltemplate repository. That’s how open-source software is improved over time.