Dear,
I am currently trying to perform so-called ‘Adiabatic relaxation simulations’ in which the rotational relaxation process of a (in my case) di-atomic Hydrogen molecule can be studied.
The main procedure of such a simulation is as follows:
- Build a periodic cell of L x L x L dimensions and fill with N molecules
- Initialize velocities by sampling from the equilibrium distribution corresponding to a specified equilibrium temperature T_inf.
- Equilibrate the system shortly (to make sure that the velocities are distributed correctly and energies are equipartitioned)
- Disturb the balance between translational and rotational energy
- Let the system relax again
I have been able to perform steps 1-4 and I verified that the system is in equilibrium and the energies are equipartitioned. I compute the different energies using the velocities of the atoms. I compute translational energy using the average of the velocities of two atoms of the same molecule. Rotational velocities and energy is computed by decomposing the velocity vectors in normal and (2) tangential components respectively.
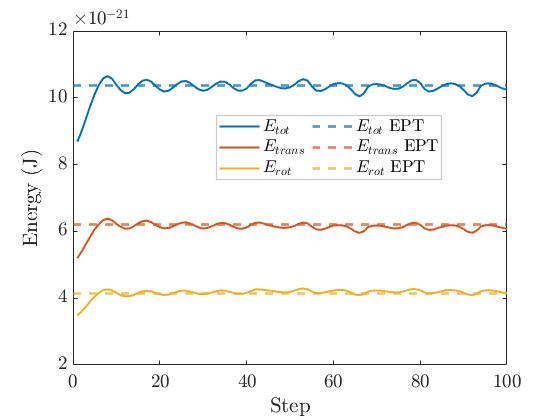
Now my aim is to re-scale the CoM velocity and angular velocities separately such that the equilibrium is disturbed. To my knowledge, LAMMPS works with atomic velocities rather than with specific angular velocities.
My main question is: Does LAMMPS have features/commands to assign/scale translational (CoM) and rotational velocities separately?
If not, I suppose the one and only method to do such thing is by basically reversing my post-processing procedure, where I went from atomic velocities to CoM and angular velocities. This will probably lead to a complex system of two equations.